18.4 Nondisjunction
Chromosome Number Abnormalities
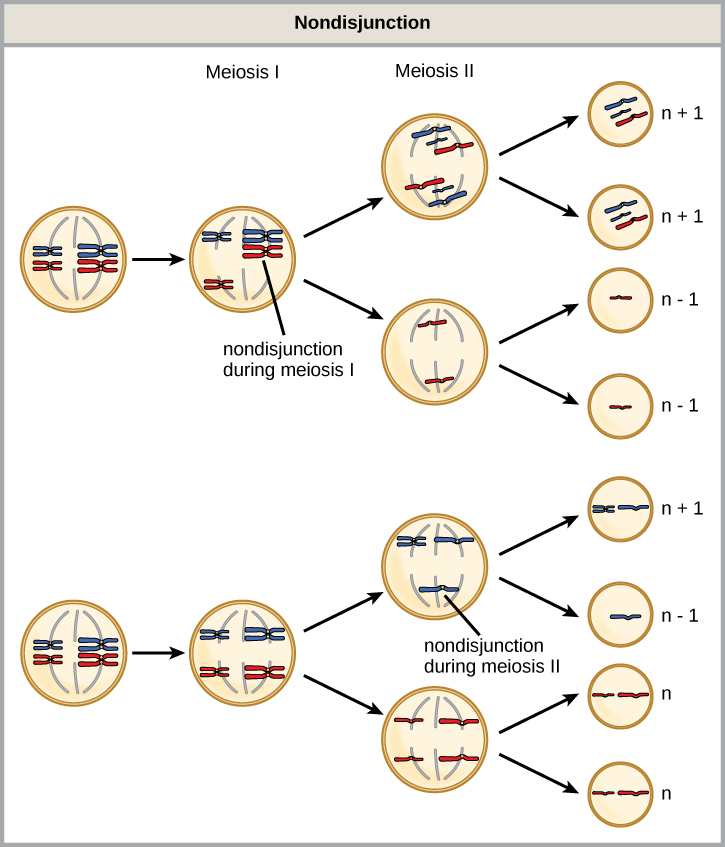
Of all of the chromosomal disorders, chromosome number abnormalities are the most obviously identifiable from a karyogram. Chromosome number disorders include duplicating or losing entire chromosomes, as well as changes in the number of complete sets of chromosomes. They are caused by nondisjunction, which occurs when homologous chromosome pairs or sister chromatids fail to separate during meiosis. Misaligned or incomplete synapsis, or a spindle apparatus dysfunction that facilitates chromosome migration, can cause nondisjunction. The risk of nondisjunction occurring increases with the parents’ age.
Nondisjunction can occur during either meiosis I or II, with differing results. If homologous chromosomes fail to separate during meiosis I, the result is two gametes that lack that particular chromosome and two gametes with two chromosome copies. If sister chromatids fail to separate during meiosis II, the result is one gamete that lacks that chromosome, two normal gametes with one chromosome copy, and one gamete with two chromosome copies.
Aneuploidy
Scientists call an individual in which every chromosome is part of a complete set euploid. Thus haploid, diploid, triploid, tetraploid, etc. individuals are euploid. In humans, euploidy corresponds to 22 pairs of autosomes and one pair of sex chromosomes. An individual with an abnormal copy number of one chromosome type is described as aneuploid, a term that includes monosomy (missing one chromosome) or trisomy (possessing an extraneous chromosome). Monosomic human zygotes missing any one copy of an autosome invariably fail to develop to birth because of a reduced copy number of a large set of genes (remember that an average human chromosome contains approximately 1000 genes). This underscores the importance of “gene dosage” in humans. Most autosomal trisomies also fail to develop to birth; however, duplications of some smaller chromosomes (13, 15, 18, 21, or 22 ) can result in offspring that survive for several weeks to many years. Trisomic individuals suffer from a different type of genetic imbalance: an excess in gene dose. Individuals with an extra chromosome may synthesize an abundance of the gene products, which that chromosome encodes. This extra dose (150 percent) of specific genes can lead to a number of functional challenges and often precludes development. The most common trisomy among viable births is that of chromosome 21, which corresponds to Down Syndrome.
Sex Chromosome Nondisjunction in Humans
Humans display dramatic deleterious effects with autosomal trisomies and monosomies. Therefore, it may seem counterintuitive that human females and males can function normally, despite carrying different numbers of the X chromosome. (Note that when we use the words “female” and “male” in this textbook, we are referring to biological sex, not gender. Although gender and sex are related, sex is a biological concept, while gender is based on how a person identifies).
Rather than a gain or loss of autosomes, variations in the number of sex chromosomes occur with no effects, or with relatively mild effects. In part, this happens because of the molecular process of X inactivation. Early in development, when female mammalian embryos consist of just a few thousand cells (relative to trillions in the newborn), one X chromosome in each cell inactivates by tightly condensing into a quiescent (dormant) structure, called a Barr body. The chance that an X chromosome (maternally or paternally derived) inactivates in each cell is random, but once this occurs, all cells derived from that one will have the same inactive X chromosome or Barr body. By this process, females compensate for their double genetic dose of X chromosome.
An individual carrying an abnormal number of X chromosomes will inactivate all but one X chromosome in each of their cells. However, even inactivated X chromosomes continue to express a few genes, and X chromosomes must reactivate for the proper maturation of female ovaries. As a result, X-chromosomal abnormalities typically occur with mild mental and physical defects, as well as sterility. If the X chromosome is absent altogether, the individual will not develop in utero.
This takes place in other mammals as well. For example, in “tortoiseshell” cats, we observe embryonic X inactivation as color variegation. Females that are heterozygous for an X-linked coat color gene will express one of two different coat colors over different regions of their body, corresponding to whichever X chromosome inactivates in that region’s embryonic cell progenitor.

Scientists have identified and characterized several variants in sex chromosome number, including XXX, XXY, XYY, and X0. Individuals with three X chromosomes are phenotypically female. They are often phenotypically normal, but sometimes express developmental delays and reduced fertility. The XXY genotype, corresponding to one type of Klinefelter syndrome, corresponds to phenotypically male individuals. Often they have no symptoms except for infertility, or very subtle symptoms. Sometimes this disorder is associated with small testes, enlarged breasts, and reduced body hair. More complex types of Klinefelter syndrome exist in which the individual has as many as five X chromosomes. In all types, every X chromosome except one undergoes inactivation to compensate for the excess genetic dosage. We see this as several Barr bodies in each cell nucleus. XYY individual are phenotypically male, and usually have few or no symptoms, although there is an increased risk of learning disabilities. Turner syndrome, characterized as an X0 genotype (i.e., only a single X chromosome and no Y chromosome), corresponds to a phenotypically female individual. Symptoms vary, but can include short stature, webbed skin in the neck region, hearing and cardiac impairments, and sterility.
