9 Chapter 9: DNA Structure, Protein Synthesis and GMO’s
Chapter Outline
- 9.1 DNA Structure
- 9.2 Basics of DNA Replication
- 9.3 DNA Replication in Prokaryotes
- 9.4 DNA Replication in Eukaryotes
- 9.5 DNA Repair
- 9.6 Genetic Code
- 9.7 Prokaryotic Transcription
- 9.8 Eurkaryotic Transcription
- 9.9 RNA Processing in Eukaryotes
- 9.10 Ribosomes and Protein Synthesis
- 9.11 Regulation of Gene Expression
- 9.12 Prokaryotic Gene Regulation
- 9.13 Eukaryotic Gene Regulation
- 9.14 Eukaryotic Transcription Gene Regulation
- 9.15 Eukaryotic Post-Transcriptional Gene Regulation
- 9.16 Eukaryotic Translational and Post-Translational Gene Regulation
- 9.17 Cancer and Gene Regulation
- 9.18 Biotechnology
- 9.19 Mapping Genomes
- 9.20 Whole-Genome Sequencing
- 9.21 Applying Genomics
- 9.22 Genomics and Proteomics

Figure 9.1 Dolly the sheep was the first large mammal to be cloned.
Introduction
The three letters “DNA” have now become synonymous with crime solving, paternity testing, human identification, and genetic testing. DNA can be retrieved from hair, blood, or saliva. Each person’s DNA is unique, and it is possible to detect differences between individuals within a species on the basis of these unique features.
Whereas each cell shares the same genome and DNA sequence, each cell does not turn on, or express, the same set of genes. Each cell type needs a different set of proteins to perform its function. Therefore, only a small subset of proteins is expressed in a cell. For the proteins to be expressed, the DNA must be transcribed into RNA and the RNA must be translated into protein. In a given cell type, not all genes encoded in the DNA are transcribed into RNA or translated into protein because specific cells in our body have specific functions. Specialized proteins that make up the eye (iris, lens, and cornea) are only expressed in the eye, whereas the specialized proteins in the heart (pacemaker cells, heart muscle, and valves) are only expressed in the heart. At any given time, only a subset of all of the genes encoded by our DNA are expressed and translated into proteins. The expression of specific genes is a highly regulated process with many levels and stages of control. This complexity ensures the proper expression in the proper cell at the proper time.
Since the rediscovery of Mendel’s work in 1900, the definition of the gene has progressed from an abstract unit of heredity to a tangible molecular entity capable of replication, expression, and mutation. Genes are composed of DNA and are linearly arranged on chromosomes. Genes specify the sequences of amino acids, which are the building blocks of proteins. In turn, proteins are responsible for orchestrating nearly every function of the cell. Both genes and the proteins they encode are absolutely essential to life as we know it.
Learning Objectives
You will be able to describe the structure and function of DNA and how it is translated into proteins:
- Explain how DNA is copied to carry the information of heredity
- Describe how DNA is transcribed and translated into proteins:
- Explain how genes in DNA code for proteins
- Identify or diagram how information flows from DNA to protein
- Recognize the process of transcription to make a mRNA from DNA
- Recognize the process of translation to “read” mRNA codons to make a protein
9.1 | DNA Structure
The building blocks of DNA are nucleotides. The important components of the nucleotide are a nitrogenous base, deoxyribose (5-carbon sugar), and a phosphate group (Figure 9.5). The nucleotide is named depending on the nitrogenous base. The nitrogenous base can be a purine such as adenine (A) and guanine (G), or a pyrimidine such as cytosine (C) and thymine (T).
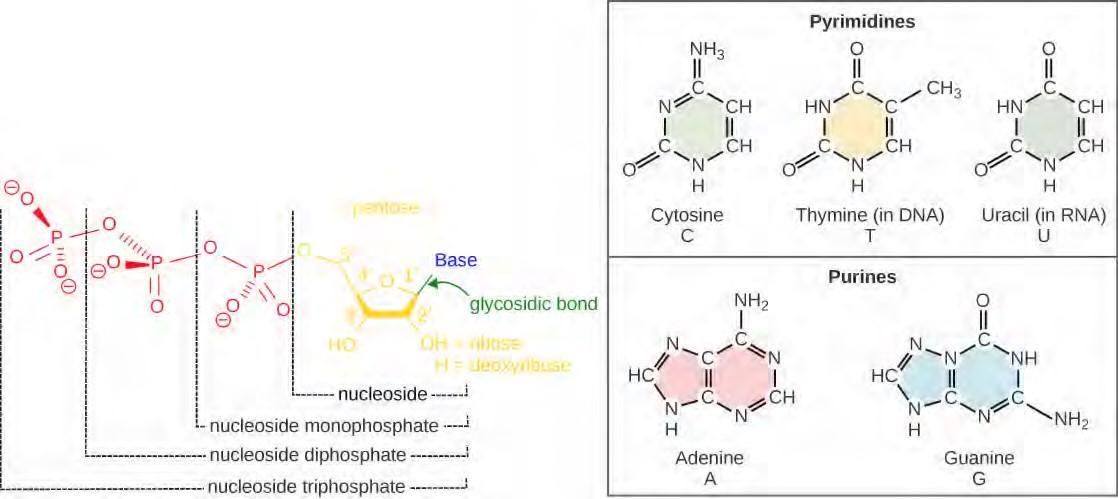
Figure 9.5 Each nucleotide is made up of a sugar, a phosphate group, and a nitrogenous base. The sugar is deoxyribose in DNA and ribose in RNA.
In the 1950s, Francis Crick and James Watson worked together to determine the structure of DNA at the University of Cambridge, England. Other scientists like Linus Pauling and Maurice Wilkins were also actively exploring this field. Pauling had discovered the secondary structure of proteins using X-ray crystallography. In Wilkins’ lab, researcher Rosalind Franklin was using X-ray diffraction methods to understand the structure of DNA. Watson and Crick were able to piece together the puzzle of the DNA molecule on the basis of Franklin’s data because Crick had also studied X-ray diffraction (Figure 9.6). In 1962, James Watson, Francis Crick, and Maurice Wilkins were awarded the Nobel Prize in Medicine. Unfortunately, by then Franklin had died, and Nobel prizes are not awarded posthumously.

Figure 9.6 The work of pioneering scientists (a) James Watson, Francis Crick, and Maclyn McCarty led to our present day understanding of DNA. Scientist Rosalind Franklin discovered (b) the X-ray diffraction pattern of DNA, which helped to elucidate its double helix structure. (credit a: modification of work by Marjorie McCarty, Public Library of Science)
Watson and Crick proposed that DNA is made up of two strands that are twisted around each other to form a right-handed helix. Base pairing takes place between a purine and pyrimidine; namely, A pairs with T and G pairs with C. Adenine and thymine are complementary base pairs, and cytosine and guanine are also complementary base pairs. The base pairs are stabilized by hydrogen bonds; adenine and thymine form two hydrogen bonds and cytosine and guanine form three hydrogen bonds. The two strands are anti-parallel in nature; that is, the 3′ end of one strand faces the 5′ end of the other strand. The sugar and phosphate of the nucleotides form the backbone of the structure, whereas the nitrogenous bases are stacked inside. (Figure 9.7).
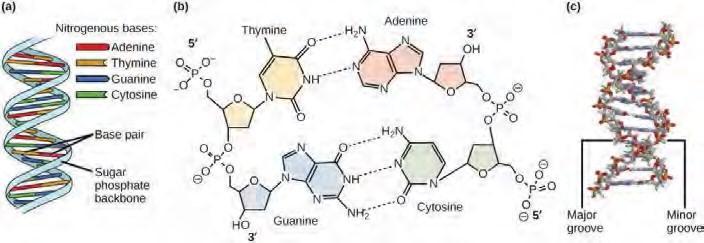
Figure 9.7 DNA has (a) a double helix structure and (b) phosphodiester bonds. The (c) major and minor grooves are binding sites for DNA binding proteins during processes such as transcription (the copying of RNA from DNA) and replication.
Gel electrophoresis is a technique used to separate DNA fragments of different sizes. Usually the gel is made of a chemical called agarose. The DNA has a net negative charge and moves from the negative electrode toward the positive electrode. The electric current is applied for sufficient time to let the DNA separate according to size; the smallest fragments will be farthest from the well (where the DNA was loaded), and the heavier molecular weight fragments will be closest to the well. Once the DNA is separated, the gel is stained with a DNA-specific dye for viewing it (Figure 9.9).
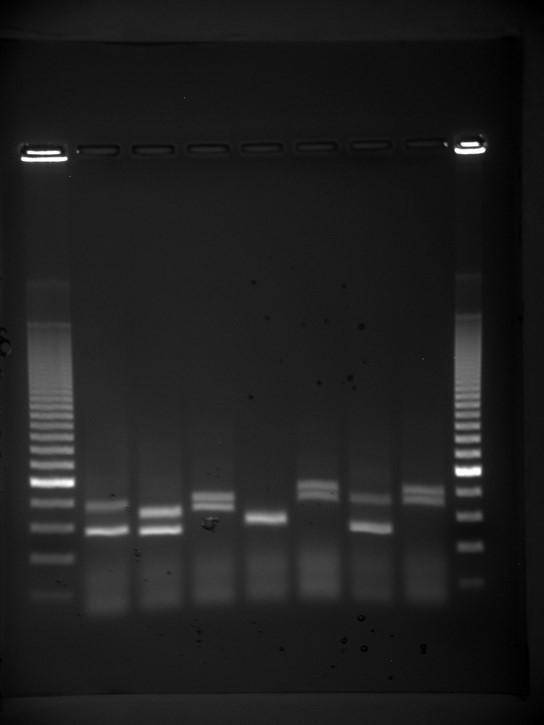
Figure 9.9 DNA can be separated on the basis of size using gel electrophoresis. (credit: James Jacob, Tompkins Cortland Community College)

Watch Svante Pääbo’s talk (http://openstaxcollege.org/l/neanderthal) explaining the Neanderthal genome research at the 2011 annual TED (Technology, Entertainment, Design) conference.
DNA Packaging in Cells
When comparing prokaryotic cells to eukaryotic cells, prokaryotes are much simpler than eukaryotes in many of their features (Figure 9.10). Most prokaryotes contain a single, circular chromosome that is found in an area of the cytoplasm called the nucleoid.
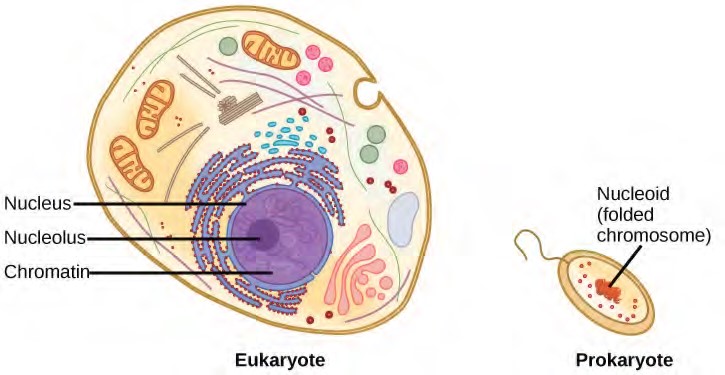
The size of the genome in one of the most well-studied prokaryotes, E.coli, is 4.6 million base pairs (approximately 1.1 mm, if cut and stretched out). So how does this fit inside a small bacterial cell? The DNA is twisted by what is known as supercoiling. Supercoiling means that DNA is either under-wound (less than one turn of the helix per 10 base pairs) or over-wound (more than 1 turn per 10 base pairs) from its normal relaxed state. Some proteins are known to be involved in the supercoiling; other proteins and enzymes such as DNA gyrase help in maintaining the supercoiled structure.
Eukaryotes, whose chromosomes each consist of a linear DNA molecule, employ a different type of packing strategy to fit their DNA inside the nucleus (Figure 9.11). At the most basic level, DNA is wrapped around proteins known as histones to form structures called nucleosomes. The histones are evolutionarily conserved proteins that are rich in basic amino acids and form an octamer. The DNA (which is negatively charged because of the phosphate groups) is wrapped tightly around the histone core. This nucleosome is linked to the next one with the help of a linker DNA. This is also known as the “beads on a string” structure. This is further compacted into a 30 nm fiber, which is the diameter of the structure. At the metaphase stage, the chromosomes are at their most compact, are approximately 700 nm in width, and are found in association with scaffold proteins.In interphase, eukaryotic chromosomes have two distinct regions that can be distinguished by staining. The tightly packaged region is known as heterochromatin, and the less dense region is known as euchromatin. Heterochromatin usually contains genes that are not expressed, and is found in the regions of the centromere and telomeres. The euchromatin usually contains genes that are transcribed, with DNA packaged around nucleosomes but not further compacted.
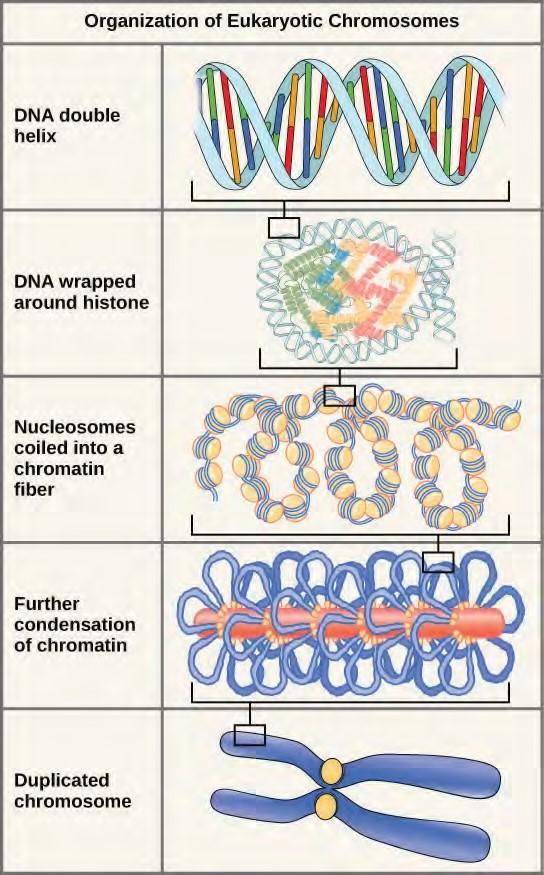
Figure 9.11 These figures illustrate the compaction of the eukaryotic chromosome.
9.2 | Basics of DNA Replication
The elucidation of the structure of the double helix provided a hint as to how DNA divides and makes copies of itself. This model suggests that the two strands of the double helix separate during replication, and each strand serves as a template from which the new complementary strand is copied. What was not clear was how the replication took place. There were three models suggested (Figure 9.12): conservative, semi-conservative, and dispersive.
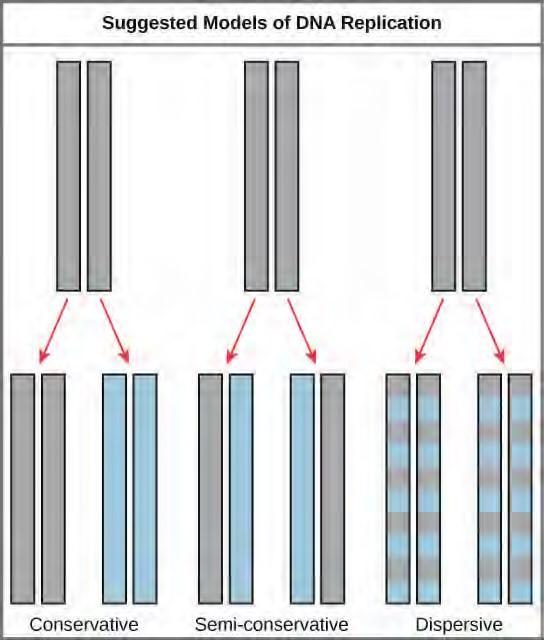
Figure 9.12 The three suggested models of DNA replication. Grey indicates the original DNA strands, and blue indicates newly synthesized DNA.
Click through this tutorial (http://openstaxcollege.org/l/DNA_replicatio2) on DNA replication.
9.3 | DNA Replication in Prokaryotes
DNA replication has been extremely well studied in prokaryotes primarily because of the small size of the genome and the mutants that are available. E. coli has 4.6 million base pairs in a single circular chromosome and all of it gets replicated in approximately 42 minutes, starting from a single origin of replication and proceeding around the circle in both directions. This means that approximately 1000 nucleotides are added per second. The process is quite rapid and occurs without many mistakes.
How does the replication machinery know where to begin? It turns out that there are specific nucleotide sequences called origins of replication where replication begins. In E. coli, which has a single origin of replication on its one chromosome (as do most prokaryotes), it is approximately 245 base pairs long and is rich in AT sequences. The origin of replication is recognized by certain proteins that bind to this site. An enzyme called helicase unwinds the DNA by breaking the hydrogen bonds between the nitrogenous base pairs. ATP hydrolysis is required for this process. As the DNA opens up, Y-shaped structures called replication forks are formed. Two replication forks are formed at the origin of replication and these get extended bi- directionally as replication proceeds. Single-strand binding proteins coat the single strands of DNA near the replication fork to prevent the single-stranded DNA from winding back into a double helix. DNA polymerase is able to add nucleotides only in the 5′ to 3′ direction (a new DNA strand can be only extended in this direction). It also requires a free 3′-OH group to which it can add nucleotides by forming a phosphodiester bond between the 3′-OH end and the 5′ phosphate of the next nucleotide. This essentially means that it cannot add nucleotides if a free 3′-OH group is not available. Then how does it add the first nucleotide? The problem is solved with the help of a primer that provides the free 3′-OH end. Another enzyme, RNA primase, synthesizes an RNA primer that is about five to ten nucleotides long and complementary to the DNA. Because this sequence primes the DNA synthesis, it is appropriately called the primer. DNA polymerase can now extend this RNA primer, adding nucleotides one by one that are complementary to the template strand (Figure 9.14).
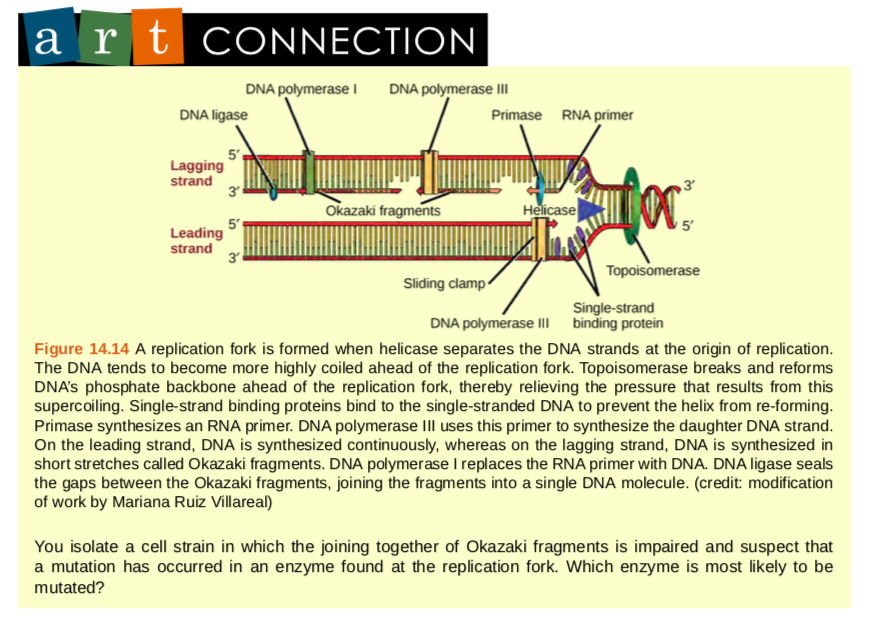
The replication fork moves at the rate of 1000 nucleotides per second. DNA polymerase can only extend in the 5′ to 3′ direction, which poses a slight problem at the replication fork. As we know, the DNA double helix is anti-parallel; that is, one strand is in the 5′ to 3′ direction and the other is oriented in the 3′ to 5′ direction. One strand, which is complementary to the 3′ to 5′ parental DNA strand, is synthesized continuously towards the replication fork because the polymerase can add nucleotides in this direction. This continuously synthesized strand is known as the leading strand. The other strand, complementary to the 5′ to 3′ parental DNA, is extended away from the replication fork, in small fragments known as Okazaki fragments, each requiring a primer to start the synthesis. Okazaki fragments are named after the Japanese scientist who first discovered them. The strand with the Okazaki fragments is known as the lagging strand.The leading strand can be extended by one primer alone, whereas the lagging strand needs a new primer for each of the short Okazaki fragments. The overall direction of the lagging strand will be 3′ to 5′, and that of the leading strand 5′ to 3′. A protein called the sliding clamp holds the DNA polymerase in place as it continues to add nucleotides. The sliding clamp is a ring-shaped protein that binds to the DNA and holds the polymerase in place. Topoisomerase prevents the over-winding of the DNA double helix ahead of the replication fork as the DNA is opening up; it does so by causing temporary nicks in the DNA helix and then resealing it. As synthesis proceeds, the RNA primers are replaced by DNA. The primers are removed by the exonuclease activity of DNA pol I, and the gaps are filled in by deoxyribonucleotides. The nicks that remain between the newly synthesized DNA (that replaced the RNA primer) and the previously synthesized DNA are sealed by the enzyme DNA ligase that catalyzes the formation of phosphodiester linkage between the 3′-OH end of one nucleotide and the 5′ phosphate end of the other fragment. Once the chromosome has been completely replicated, the two DNA copies move into two different cells during cell division.
The process of DNA replication can be summarized as follows:
- DNA unwinds at the origin of replication.
- Helicase opens up the DNA-forming replication forks; these are extended bidirectionally.
- Single-strand binding proteins coat the DNA around the replication fork to prevent rewinding of the DNA.
- Topoisomerase binds at the region ahead of the replication fork to prevent supercoiling.
- Primase synthesizes RNA primers complementary to the DNA strand.
- DNA polymerase starts adding nucleotides to the 3′-OH end of the primer.
- Elongation of both the lagging and the leading strand continues.
- RNA primers are removed by exonuclease activity.
- Gaps are filled by DNA pol by adding dNTPs.
- The gap between the two DNA fragments is sealed by DNA ligase, which helps in the formation of phosphodiester bonds.
Table 9.1 summarizes the enzymes involved in prokaryotic DNA replication and the functions of each.
Prokaryotic DNA Replication: Enzymes and Their Function
|
Enzyme/protein |
Specific Function |
|
DNA pol I |
Exonuclease activity removes RNA primer and replaces with newly synthesized DNA |
|
DNA pol II |
Repair function |
|
DNA pol III |
Main enzyme that adds nucleotides in the 5′-3′ direction |
|
Helicase |
Opens the DNA helix by breaking hydrogen bonds between the nitrogenous bases |
|
Ligase |
Seals the gaps between the Okazaki fragments to create one continuous DNA strand |
|
Primase |
Synthesizes RNA primers needed to start replication |
|
Sliding Clamp |
Helps to hold the DNA polymerase in place when nucleotides are being added |
|
Topoisomerase |
Helps relieve the stress on DNA when unwinding by causing breaks and then resealing the DNA |
|
Single-strand binding proteins (SSB) |
Binds to single-stranded DNA to avoid DNA rewinding back. |
Review the full process of DNA replication here (http://openstaxcollege.org/l/replication_DNA) .
9.4 | DNA Replication in Eukaryotes
Eukaryotic genomes are much more complex and larger in size than prokaryotic genomes. The human genome has three billion base pairs per haploid set of chromosomes, and 6 billion base pairs are replicated during the S phase of the cell cycle. There are multiple origins of replication on the eukaryotic chromosome; humans can have up to 100,000 origins of replication. The rate of replication is approximately 100 nucleotides per second, much slower than prokaryotic replication. In yeast, which is a eukaryote, special sequences known as Autonomously Replicating Sequences (ARS) are found on the chromosomes. These are equivalent to the origin of replication in E. coli.
A helicase using the energy from ATP hydrolysis opens up the DNA helix. Replication forks are formed at each replication origin as the DNA unwinds. The opening of the double helix causes overwinding, or supercoiling, in the DNA ahead of the replication fork. These are resolved with the action of topoisomerases. Primers are formed by the enzyme primase, and using the primer, DNA pol can start synthesis. While the leading strand is continuously synthesized by the enzyme pol δ, the lagging strand is synthesized by pol ε. A sliding clamp protein known as PCNA (Proliferating Cell Nuclear Antigen) holds the DNA pol in place so that it does not slide off the DNA. RNase H removes the RNA primer, which is then replaced with DNA nucleotides. The Okazaki fragments in the lagging strand are joined together after the replacement of the RNA primers with DNA. The gaps that remain are sealed by DNA ligase, which forms the phosphodiester bond.
Telomere replication
Unlike prokaryotic chromosomes, eukaryotic chromosomes are linear. As you’ve learned, the enzyme DNA pol can add nucleotides only in the 5′ to 3′ direction. In the leading strand, synthesis continues until the end of the chromosome is reached. On the lagging strand, DNA is synthesized in short stretches, each of which is initiated by a separate primer. When the replication fork reaches the end of the linear chromosome, there is no place for a primer to be made for the DNA fragment to be copied at the end of the chromosome. These ends thus remain unpaired, and over time these ends may get progressively shorter as cells continue to divide.
The ends of the linear chromosomes are known as telomeres, which have repetitive sequences that code for no particular gene. In a way, these telomeres protect the genes from getting deleted as cells continue to divide. In humans, a six base pair sequence, TTAGGG, is repeated 100 to 1000 times. The discovery of the enzyme telomerase (Figure 14.16) helped in the understanding of how chromosome ends are maintained. The telomerase enzyme contains a catalytic part and a built-in RNA template. It attaches to the end of the chromosome, and complementary bases to the RNA template are added on the 3′ end of the DNA strand. Once the 3′ end of the lagging strand template is sufficiently elongated, DNA polymerase can add the nucleotides complementary to the ends of the chromosomes. Thus, the ends of the chromosomes are replicated.
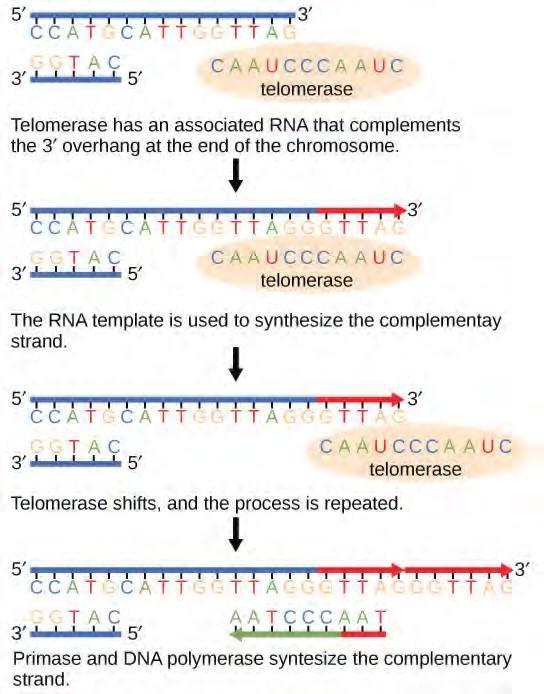
Figure 9.15 The ends of linear chromosomes are maintained by the action of the telomerase enzyme.Telomerase is typically active in germ cells and adult stem cells. It is not active in adult somatic cells. For her discovery of telomerase and its action, Elizabeth Blackburn (Figure 9.16) received the Nobel Prize for Medicine and Physiology in 2009.

Figure 9.16 Elizabeth Blackburn, 2009 Nobel Laureate, is the scientist who discovered how telomerase works. (credit: US Embassy Sweden)
Telomerase and Aging
Cells that undergo cell division continue to have their telomeres shortened because most somatic cells do not make telomerase. This essentially means that telomere shortening is associated with aging. With the advent of modern medicine, preventative health care, and healthier lifestyles, the human life span has increased, and there is an increasing demand for people to look younger and have a better quality of life as they grow older. In 2010, scientists found that telomerase can reverse some age-related conditions in mice. This may have potential in regenerative medicine. Telomerase-deficient mice were used in these studies; these mice have tissue atrophy, stem cell depletion, organ system failure, and impaired tissue injury responses [2]. Telomerase reactivation in these mice caused extension of telomeres, reduced DNA damage, reversed neurodegeneration, and improved the function of the testes, spleen, and intestines. Thus, telomere reactivation may have potential for treating age-related diseases in humans.
2. Jaskelioff et al., “Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice,” Nature 469 (2011): 102-7.
Difference between Prokaryotic and Eukaryotic Replication
|
Property |
Prokaryotes |
Eukaryotes |
|
Origin of replication |
Single |
Multiple |
|
Rate of replication |
1000 nucleotides/s |
50 to 100 nucleotides/s |
|
DNA polymerase types |
5 |
14 |
|
Telomerase |
Not present |
Present |
|
RNA primer removal |
DNA pol I |
RNase H |
|
Strand elongation |
DNA pol III |
Pol δ, pol ε |
|
Sliding clamp |
Sliding clamp |
PCNA |
Table 9.2
9.5 | DNA Repair
DNA replication is a highly accurate process, but mistakes can occasionally occur, such as a DNA polymerase inserting a wrong base. Uncorrected mistakes may sometimes lead to serious consequences, such as cancer. Repair mechanisms correct the mistakes. In rare cases, mistakes are not corrected, leading to mutations; in other cases, repair enzymes are themselves mutated or defective.
Most of the mistakes during DNA replication are promptly corrected by DNA polymerase by proofreading the base that has been just added (Figure 9.17). In proofreading, the DNA pol reads the newly added base before adding the next one, so a correction can be made. The polymerase checks whether the newly added base has paired correctly with the base in the template strand. If it is the right base, the next nucleotide is added. If an incorrect base has been added, the enzyme makes a cut at the phosphodiester bond and releases the wrong nucleotide. This is performed by the exonuclease action of DNA pol III. Once the incorrect nucleotide has been removed, a new one will be added again.
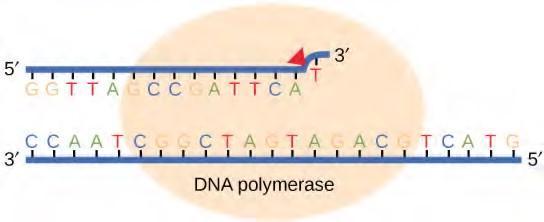
Figure 9.17 Proofreading by DNA polymerase corrects errors during replication.
Some errors are not corrected during replication, but are instead corrected after replication is completed; this type of repair is known as mismatch repair (Figure 9.18). The enzymes recognize the incorrectly added nucleotide and excise it; this is then replaced by the correct base. If this remains uncorrected, it may lead to more permanent damage. How do mismatch repair enzymes recognize which of the two bases is the incorrect one? In E. coli, after replication, the nitrogenous base adenine acquires a methyl group; the parental DNA strand will have methyl groups, whereas the newly synthesized strand lacks them. Thus, DNA polymerase is able to remove the wrongly incorporated bases from the newly synthesized, non-methylated strand. In eukaryotes, the mechanism is not very well understood, but it is believed to involve recognition of unsealed nicks in the new strand, as well as a short-term continuing association of some of the replication proteins with the new daughter strand after replication has completed.
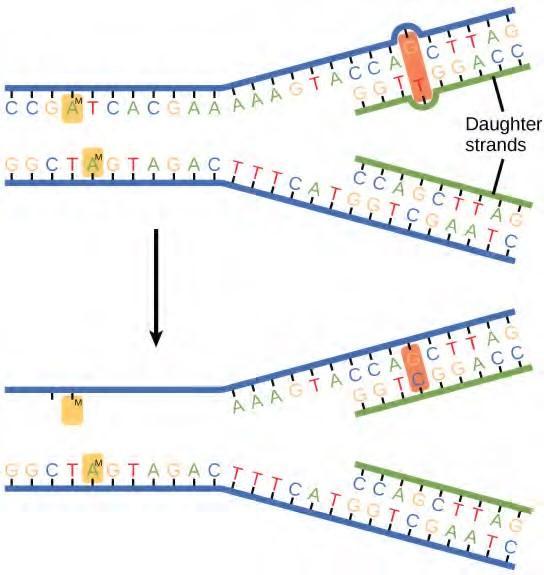
Figure 9.18 In mismatch repair, the incorrectly added base is detected after replication. The mismatch repair proteins detect this base and remove it from the newly synthesized strand by nuclease action. The gap is now filled with the correctly paired base.
In another type of repair mechanism, nucleotide excision repair, enzymes replace incorrect bases by making a cut on both the 3′ and 5′ ends of the incorrect base (Figure 9.19). The segment of DNA is removed and replaced with the correctly paired nucleotides by the action of DNA pol. Once the bases are filled in, the remaining gap is sealed with a phosphodiester linkage catalyzed by DNA ligase. This repair mechanism is often employed when UV exposure causes the formation of pyrimidine dimers.
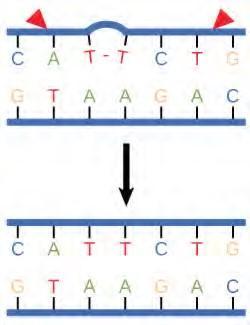
Figure 9.19 Nucleotide excision repairs thymine dimers. When exposed to UV, thymines lying adjacent to each other can form thymine dimers. In normal cells, they are excised and replaced.
A well-studied example of mistakes not being corrected is seen in people suffering from xeroderma pigmentosa (Figure 9.20). Affected individuals have skin that is highly sensitive to UV rays from the sun. When individuals are exposed to UV, pyrimidine dimers, especially those of thymine, are formed; people with xeroderma pigmentosa are not able to repair the damage. These are not repaired because of a defect in the nucleotide excision repair enzymes, whereas in normal individuals, the thymine dimers are excised and the defect is corrected. The thymine dimers distort the structure of the DNA double helix, and this may cause problems during DNA replication. People with xeroderma pigmentosa may have a higher risk of contracting skin cancer than those who don’t have the condition.
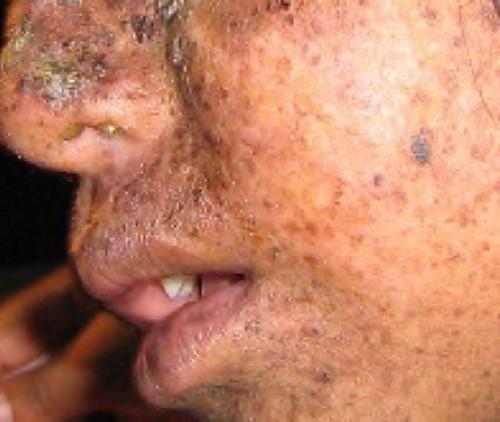
Figure 9.20 Xeroderma pigmentosa is a condition in which thymine dimerization from exposure to UV is not repaired. Exposure to sunlight results in skin lesions. (credit: James Halpern et al.)
Errors during DNA replication are not the only reason why mutations arise in DNA. Mutations, variations in the nucleotide sequence of a genome, can also occur because of damage to DNA. Such mutations may be of two types: induced or spontaneous. Induced mutations are those that result from an exposure to chemicals, UV rays, x-rays, or some other environmental agent. Spontaneous mutations occur without any exposure to any environmental agent; they are a result of natural reactions taking place within the body.
Mutations may have a wide range of effects. Some mutations are not expressed; these are known as silent mutations. Point mutations are those mutations that affect a single base pair. The most common nucleotide mutations are substitutions, in which one base is replaced by another. These can be of two types, either transitions or transversions. Transition substitution refers to a purine or pyrimidine being replaced by a base of the same kind; for example, a purine such as adenine may be replaced by the purine guanine. Transversion substitution refers to a purine being replaced by a pyrimidine, or vice versa; for example, cytosine, a pyrimidine, is replaced by adenine, a purine. Mutations can also be the result of the addition of a base, known as an insertion, or the removal of a base, also known as deletion. Sometimes a piece of DNA from one chromosome may get translocated to another chromosome or to another region of the same chromosome; this is also known as translocation. These mutation types are shown in Figure 9.21.
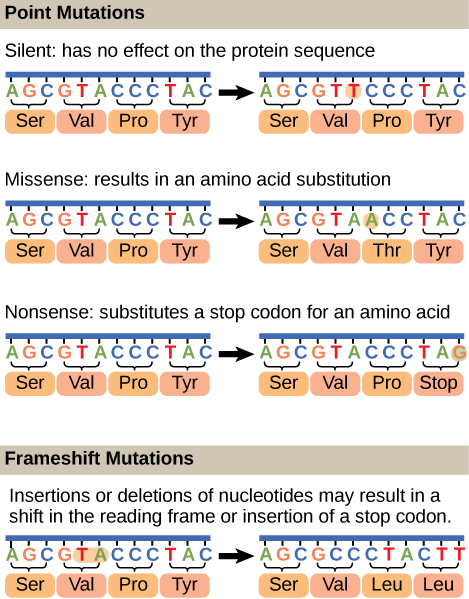
Mutations in repair genes have been known to cause cancer. Many mutated repair genes have been implicated in certain forms of pancreatic cancer, colon cancer, and colorectal cancer. Mutations can affect either somatic cells or germ cells. If many mutations accumulate in a somatic cell, they may lead to problems such as the uncontrolled cell division observed in cancer. If a mutation takes place in germ cells, the mutation will be passed on to the next generation, as in the case of hemophilia and xeroderma pigmentosa.
9.6 | The Genetic Code
The cellular process of transcription generates messenger RNA (mRNA), a mobile molecular copy of one or more genes with an alphabet of A, C, G, and uracil (U). Translation of the mRNA template converts nucleotide-based genetic information into a protein product. Protein sequences consist of 20 commonly occurring amino acids; therefore, it can be said that the protein alphabet consists of 20 letters (Figure 9.21). Each amino acid is defined by a three-nucleotide sequence called the triplet codon. Different amino acids have different chemistries (such as acidic versus basic, or polar and nonpolar) and different structural constraints. Variation in amino acid sequence gives rise to enormous variation in protein structure and function.
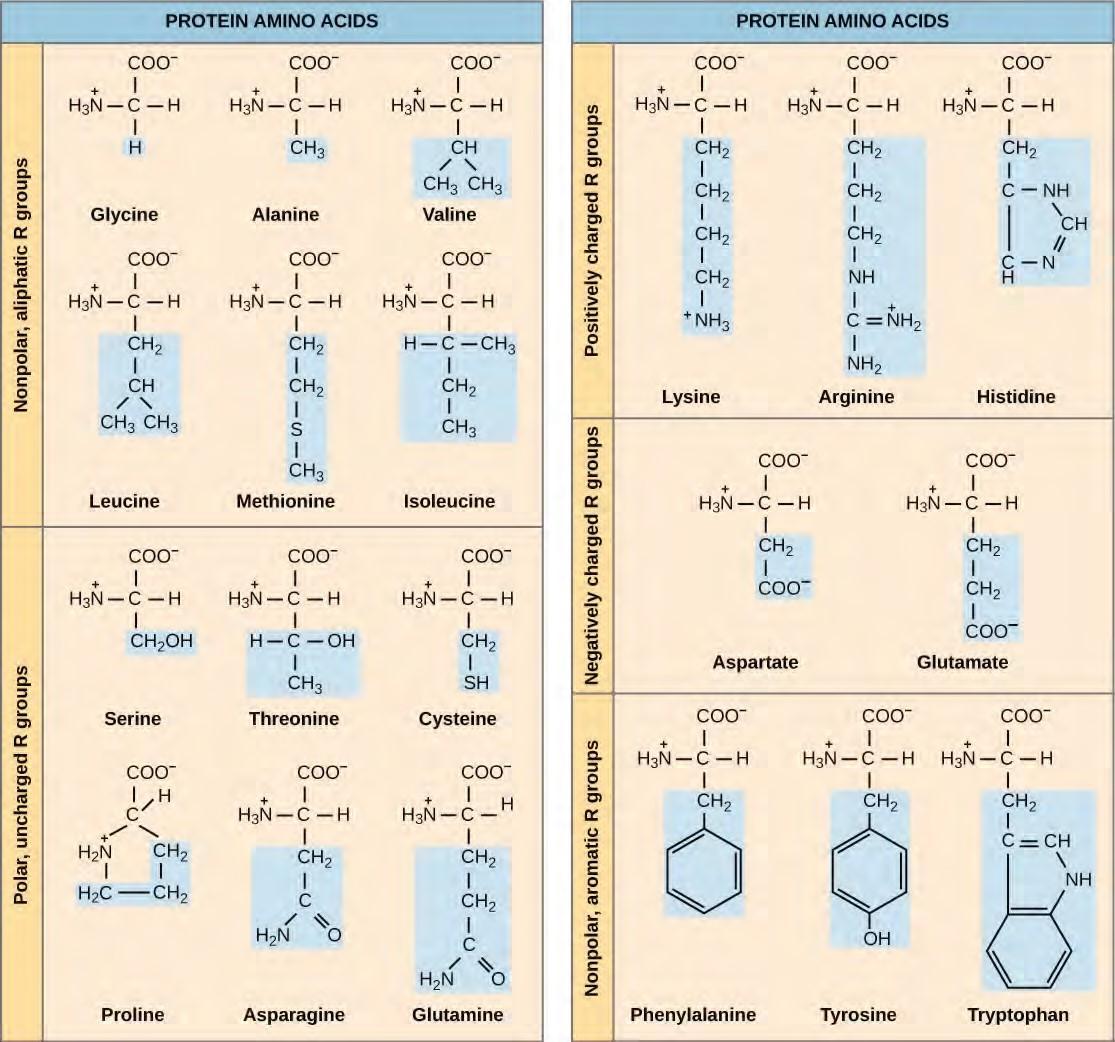
Figure 9.21 Structures of the 20 amino acids found in proteins are shown. Each amino acid is composed of an amino group ( NH+3 ), a carboxyl group (COO–), and a side chain (blue). The side chain may be nonpolar, polar, or charged, as well as large or small. It is the variety of amino acid side chains that gives rise to the incredible variation of protein structure and function.
The Central Dogma: DNA Encodes RNA; RNA Encodes Protein
The flow of genetic information in cells from DNA to mRNA to protein is described by the Central Dogma (Figure 9.22), which states that genes specify the sequence of mRNAs, which in turn specify the sequence of proteins. The decoding of one molecule to another is performed by specific proteins and RNAs. Because the information stored in DNA is so central to cellular function, it makes intuitive sense that the cell would make mRNA copies of this information for protein synthesis, while keeping the DNA itself intact and protected. The copying of DNA to RNA is relatively straightforward, with one nucleotide being added to the mRNA strand for every nucleotide read in the DNA strand. The translation to protein is a bit more complex because three mRNA nucleotides correspond to one amino acid in the polypeptide sequence. However, the translation to protein is still systematic and colinear, such that nucleotides 1 to 3 correspond to amino acid 1, nucleotides 4 to 6 correspond to amino acid 2, and so on.
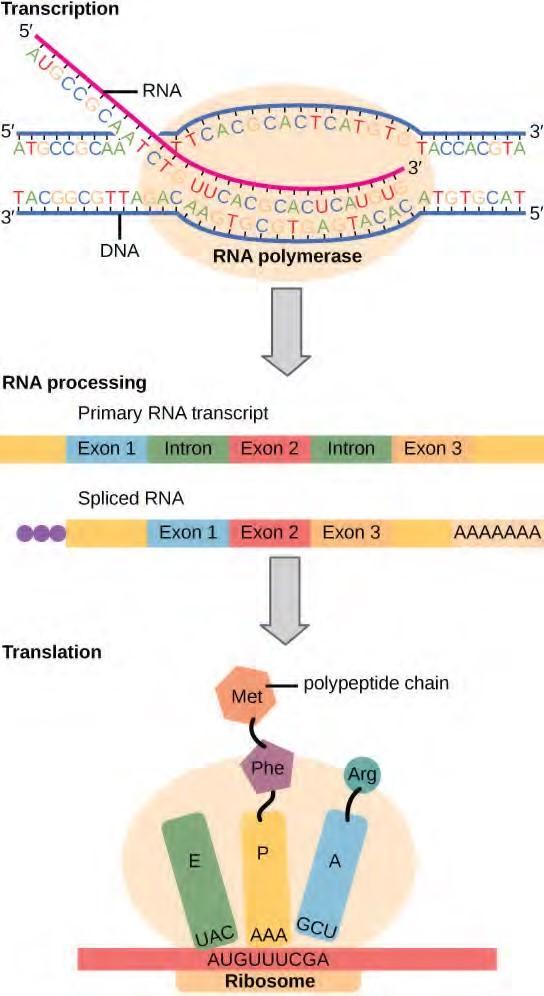
Figure 9.22 Instructions on DNA are transcribed onto messenger RNA. Ribosomes are able to read the genetic information inscribed on a strand of messenger RNA and use this information to string amino acids together into a protein.
The Genetic Code Is Degenerate and Universal
Given the different numbers of “letters” in the mRNA and protein “alphabets,” scientists theorized that combinations of nucleotides corresponded to single amino acids. Nucleotide doublets would not be sufficient to specify every amino acid because there are only 16 possible two-nucleotide combinations(42).
In contrast, there are 64 possible nucleotide triplets (43), which is far more than the number of amino acids. Scientists theorized that amino acids were encoded by nucleotide triplets and that the genetic code was degenerate. In other words, a given amino acid could be encoded by more than one nucleotide triplet. This was later confirmed experimentally; Francis Crick and Sydney Brenner used the chemical mutagen proflavin to insert one, two, or three nucleotides into the gene of a virus. When one or two nucleotides were inserted, protein synthesis was completely abolished. When three nucleotides were inserted, the protein was synthesized and functional. This demonstrated that three nucleotides specify each amino acid. These nucleotide triplets are called codons. The insertion of one or two nucleotides completely changed the triplet reading frame, thereby altering the message for every subsequent amino acid (Figure 9.24). Though insertion of three nucleotides caused an extra amino acid to be inserted during translation, the integrity of the rest of the protein was maintained.Scientists painstakingly solved the genetic code by translating synthetic mRNAs in vitro and sequencing the proteins they specified (Figure 9.23.
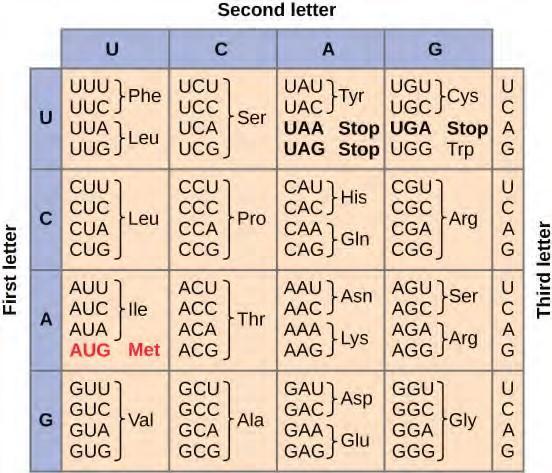
Figure 9.23 This figure shows the genetic code for translating each nucleotide triplet in mRNA into an amino acid or a termination signal in a nascent protein. (credit: modification of work by NIH)
The genetic code is universal. With a few exceptions, virtually all species use the same genetic code for protein synthesis. Conservation of codons means that a purified mRNA encoding the globin protein in horses could be transferred to a tulip cell, and the tulip would synthesize horse globin. That there is only one genetic code is powerful evidence that all of life on Earth shares a common origin, especially considering that there are about 1084possible combinations of 20 amino acids and 64 triplet codons.
Transcribe a gene and translate it to protein using complementary pairing and the genetic code at this site (http://openstaxcollege.org/l/create_protein) .
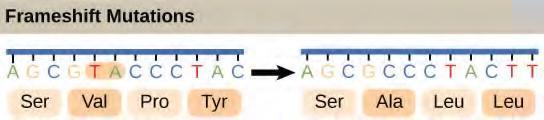
Figure 9.25 The deletion of two nucleotides shifts the reading frame of an mRNA and changes the entire protein message, creating a nonfunctional protein or terminating protein synthesis altogether.
Degeneracy is believed to be a cellular mechanism to reduce the negative impact of random mutations. Codons that specify the same amino acid typically only differ by one nucleotide. In addition, amino acids with chemically similar side chains are encoded by similar codons. This nuance of the genetic code ensures that a single-nucleotide substitution mutation might either specify the same amino acid but have no effect or specify a similar amino acid, preventing the protein from being rendered completely nonfunctional.
9.7 | Prokaryotic Transcription
The prokaryotes, which include bacteria and archaea, are mostly single-celled organisms that, by definition, lack membrane-bound nuclei and other organelles. A bacterial chromosome is a covalently closed circle that, unlike eukaryotic chromosomes, is not organized around histone proteins. The central region of the cell in which prokaryotic DNA resides is called the nucleoid. In addition, prokaryotes often have abundant plasmids, which are shorter circular DNA molecules that may only contain one or a few genes. Plasmids can be transferred independently of the bacterial chromosome during cell division and often carry traits such as antibiotic resistance.
Transcription in prokaryotes (and in eukaryotes) requires the DNA double helix to partially unwind in the region of mRNA synthesis. The region of unwinding is called a transcription bubble. Transcription always proceeds from the same DNA strand for each gene, which is called the template strand. The mRNA product is complementary to the template strand and is almost identical to the other DNA strand, called the nontemplate strand. The only difference is that in mRNA, all of the T nucleotides are replaced with U nucleotides. In an RNA double helix, A can bind U via two hydrogen bonds, just as in A–T pairing in a DNA double helix.
The nucleotide pair in the DNA double helix that corresponds to the site from which the first 5′ mRNA nucleotide is transcribed is called the +1 site, or the initiation site. Nucleotides preceding the initiation site are given negative numbers and are designated upstream. Conversely, nucleotides following the initiation site are denoted with “+” numbering and are called downstream nucleotides.
Initiation of Transcription in Prokaryotes
Prokaryotes do not have membrane-enclosed nuclei. Therefore, the processes of transcription, translation, and mRNA degradation can all occur simultaneously. The intracellular level of a bacterial protein can quickly be amplified by multiple transcription and translation events occurring concurrently on the same DNA template. Prokaryotic transcription often covers more than one gene and produces polycistronic mRNAs that specify more than one protein.
Our discussion here will exemplify transcription by describing this process in Escherichia coli, a well-studied bacterial species. Although some differences exist between transcription in E. coli and transcription in archaea, an understanding of E. coli transcription can be applied to virtually all bacterial species.
Prokaryotic RNA Polymerase
Prokaryotes use the same RNA polymerase to transcribe all of their genes. In E. coli, the polymerase is composed of five polypeptide subunits, two of which are identical. Four of these subunits, denoted α, α, β, and β‘ comprise the polymerase core enzyme. These subunits assemble every time a gene is transcribed, and they disassemble once transcription is complete. Each subunit has a unique role; the two α-subunits are necessary to assemble the polymerase on the DNA; the β-subunit binds to the ribonucleoside triphosphate that will become part of the nascent “recently born” mRNA molecule; and the β‘ binds the DNA template strand. The fifth subunit, σ, is involved only in transcription initiation. It confers transcriptional specificity such that the polymerase begins to synthesize mRNA from an appropriate initiation site. Without σ, the core enzyme would transcribe from random sites and would produce mRNA molecules that specified protein gibberish. The polymerase comprised of all five subunits is called the holoenzyme.
Prokaryotic Promoters
A promoter is a DNA sequence onto which the transcription machinery binds and initiates transcription. In most cases, promoters exist upstream of the genes they regulate. The specific sequence of a promoter is very important because it determines whether the corresponding gene is transcribed all the time, some of the time, or infrequently. Although promoters vary among prokaryotic genomes, a few elements are conserved. At the -10 and -35 regions upstream of the initiation site, there are two promoter consensus sequences, or regions that are similar across all promoters and across various bacterial species (Figure 9.26). The -10 consensus sequence, called the -10 region, is TATAAT. The -35 sequence, TTGACA, is recognized and bound by σ. Once this interaction is made, the subunits of the core enzyme bind to the site. The A–T-rich -10 region facilitates unwinding of the DNA template, and several phosphodiester bonds are made. The transcription initiation phase ends with the production of abortive transcripts, which are polymers of approximately 10 nucleotides that are made and released.
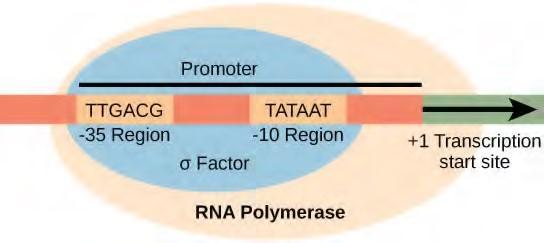
Figure 9.26 The σ subunit of prokaryotic RNA polymerase recognizes consensus sequences found in the promoter region upstream of the transcription start sight. The σ subunit dissociates from the polymerase after transcription has been initiated.
View this MolecularMovies animation (http://openstaxcollege.org/l/transcription) to see the first part of transcription and the base sequence repetition of the TATA box.
Elongation and Termination in Prokaryotes
The transcription elongation phase begins with the release of the σ subunit from the polymerase. The dissociation of σ allows the core enzyme to proceed along the DNA template, synthesizing mRNA in the 5′ to 3′ direction at a rate of approximately 40 nucleotides per second. As elongation proceeds, the DNA is continuously unwound ahead of the core enzyme and rewound behind it (Figure 9.27). The base pairing between DNA and RNA is not stable enough to maintain the stability of the mRNA synthesis components. Instead, the RNA polymerase acts as a stable linker between the DNA template and the nascent RNA strands to ensure that elongation is not interrupted prematurely.
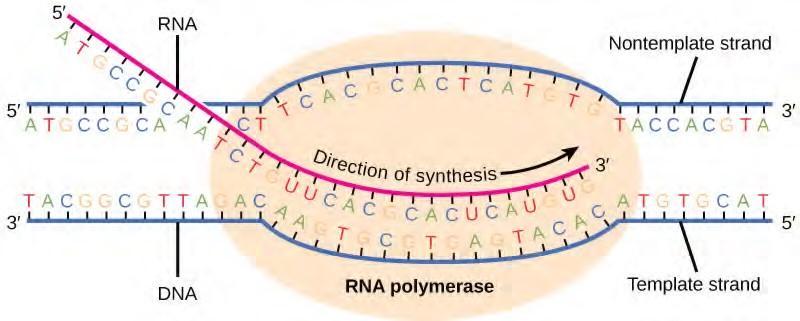
Figure 9.27 During elongation, the prokaryotic RNA polymerase tracks along the DNA template, synthesizes mRNA in the 5′ to 3′ direction, and unwinds and rewinds the DNA as it is read.
Prokaryotic Termination Signals
Once a gene is transcribed, the prokaryotic polymerase needs to be instructed to dissociate from the DNA template and liberate the newly made mRNA. Depending on the gene being transcribed, there are two kinds of termination signals. One is protein-based and the other is RNA-based. Rho-dependent termination is controlled by the rho protein, which tracks along behind the polymerase on the growing mRNA chain. Near the end of the gene, the polymerase encounters a run of G nucleotides on the DNA template and it stalls. As a result, the rho protein collides with the polymerase. The interaction with rho releases the mRNA from the transcription bubble.
Rho-independent termination is controlled by specific sequences in the DNA template strand. As the polymerase nears the end of the gene being transcribed, it encounters a region rich in C–G nucleotides. The mRNA folds back on itself, and the complementary C–G nucleotides bind together. The result is a stable hairpin that causes the polymerase to stall as soon as it begins to transcribe a region rich in A–T nucleotides. The complementary U–A region of the mRNA transcript forms only a weak interaction with the template DNA. This, coupled with the stalled polymerase, induces enough instability for the core enzyme to break away and liberate the new mRNA transcript.
Upon termination, the process of transcription is complete. By the time termination occurs, the prokaryotic transcript would already have been used to begin synthesis of numerous copies of the encoded protein because these processes can occur concurrently. The unification of transcription, translation, and even mRNA degradation is possible because all of these processes occur in the same 5′ to 3′ direction, and because there is no membranous compartmentalization in the prokaryotic cell (Figure 9.28). In contrast, the presence of a nucleus in eukaryotic cells precludes simultaneous transcription and translation.
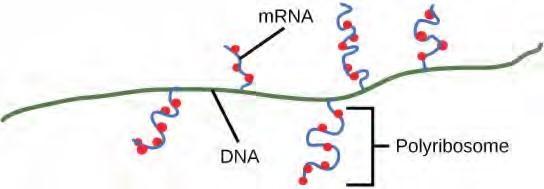
Figure 9.28 Multiple polymerases can transcribe a single bacterial gene while numerous ribosomes concurrently translate the mRNA transcripts into polypeptides. In this way, a specific protein can rapidly reach a high concentration in the bacterial cell.
Visit this BioStudio animation (http://openstaxcollege.org/l/transcription2) to see the process of prokaryotic transcription.
9.8 | Eukaryotic Transcription
Prokaryotes and eukaryotes perform fundamentally the same process of transcription, with a few key differences. The most important difference between prokaryotes and eukaryotes is the latter’s membrane bound nucleus and organelles. With the genes bound in a nucleus, the eukaryotic cell must be able to transport its mRNA to the cytoplasm and must protect its mRNA from degrading before it is translated. Eukaryotes also employ three different polymerases that each transcribe a different subset of genes. Eukaryotic mRNAs are usually monogenic, meaning that they specify a single protein.
Initiation of Transcription in Eukaryotes
Unlike the prokaryotic polymerase that can bind to a DNA template on its own, eukaryotes require several other proteins, called transcription factors, to first bind to the promoter region and then help recruit the appropriate polymerase.
The Three Eukaryotic RNA Polymerases
The features of eukaryotic mRNA synthesis are markedly more complex those of prokaryotes. Instead of a single polymerase comprising five subunits, the eukaryotes have three polymerases that are each made up of 10 subunits or more. Each eukaryotic polymerase also requires a distinct set of transcription factors to bring it to the DNA template.
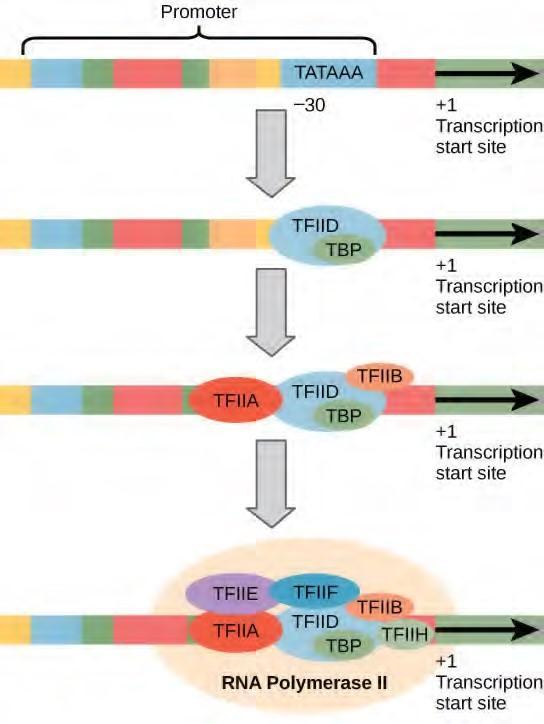
Figure 9.28 A generalized promoter of a gene transcribed by RNA polymerase II is shown. Transcription factors recognize the promoter. RNA polymerase II then binds and forms the transcription initiation complex.

Transcription Factors for RNA Polymerase II
The complexity of eukaryotic transcription does not end with the polymerases and promoters. An army of basal transcription factors, enhancers, and silencers also help to regulate the frequency with which pre-mRNA is synthesized from a gene. Enhancers and silencers affect the efficiency of transcription but are not necessary for transcription to proceed. Basal transcription factors are crucial in the formation of a preinitiation complex on the DNA template that subsequently recruits RNA polymerase II for transcription initiation.
The names of the basal transcription factors begin with “TFII” (this is the transcription factor for RNA polymerase II) and are specified with the letters A–J. The transcription factors systematically fall into place on the DNA template, with each one further stabilizing the preinitiation complex and contributing to the recruitment of RNA polymerase II.
The processes of bringing RNA polymerases I and III to the DNA template involve slightly less complex collections of transcription factors, but the general theme is the same. Eukaryotic transcription is a tightly regulated process that requires a variety of proteins to interact with each other and with the DNA strand. Although the process of transcription in eukaryotes involves a greater metabolic investment than in prokaryotes, it ensures that the cell transcribes precisely the pre-mRNAs that it needs for protein synthesis.
The Evolution of Promoters
The evolution of genes may be a familiar concept. Mutations can occur in genes during DNA replication, and the result may or may not be beneficial to the cell. By altering an enzyme, structural protein, or some other factor, the process of mutation can transform functions or physical features. However, eukaryotic promoters and other gene regulatory sequences may evolve as well. For instance, consider a gene that, over many generations, becomes more valuable to the cell. Maybe the gene encodes a structural protein that the cell needs to synthesize in abundance for a certain function. If this is the case, it would be beneficial to the cell for that gene’s promoter to recruit transcription factors more efficiently and increase gene expression. Scientists examining the evolution of promoter sequences have reported varying results. In part, this is because it is difficult to infer exactly where a eukaryotic promoter begins and ends. Some promoters occur within genes; others are located very far upstream, or even downstream, of the genes they are regulating. However, when researchers limited their examination to human core promoter sequences that were defined experimentally as sequences that bind the preinitiation complex, they found that promoters evolve even faster than protein-coding genes.
It is still unclear how promoter evolution might correspond to the evolution of humans or other higher organisms. However, the evolution of a promoter to effectively make more or less of a given gene product is an intriguing alternative to the evolution of the genes
1.H Liang et al., “Fast evolution of core promoters in primate genomes,” Molecular Biology and Evolution 25 (2008): 1239–44.
Eukaryotic Elongation and Termination
Following the formation of the preinitiation complex, the polymerase is released from the other transcription factors, and elongation is allowed to proceed as it does in prokaryotes with the polymerase synthesizing pre-mRNA in the 5′ to 3′ direction. As discussed previously, RNA polymerase II transcribes the major share of eukaryotic genes, so this section will focus on how this polymerase accomplishes elongation and termination.
Although the enzymatic process of elongation is essentially the same in eukaryotes and prokaryotes, the DNA template is more complex. When eukaryotic cells are not dividing, their genes exist as a diffuse mass of DNA and proteins called chromatin. The DNA is tightly packaged around charged histone proteins at repeated intervals. These DNA–histone complexes, collectively called nucleosomes, are regularly spaced and include 146 nucleotides of DNA wound around eight histones like thread around a spool.
For polynucleotide synthesis to occur, the transcription machinery needs to move histones out of the way every time it encounters a nucleosome. This is accomplished by a special protein complex called FACT, which stands for “facilitates chromatin transcription.” This complex pulls histones away from the DNA template as the polymerase moves along it. Once the pre-mRNA is synthesized, the FACT complex replaces the histones to recreate the nucleosomes.
The termination of transcription is different for the different polymerases. Unlike in prokaryotes, elongation by RNA polymerase II in eukaryotes takes place 1,000–2,000 nucleotides beyond the end of the gene being transcribed. This pre-mRNA tail is subsequently removed by cleavage during mRNA processing. On the other hand, RNA polymerases I and III require termination signals. Genes transcribed by RNA polymerase I contain a specific 18-nucleotide sequence that is recognized by a termination protein. The process of termination in RNA polymerase III involves an mRNA hairpin similar to rho independent termination of transcription in prokaryotes.
9.9 | RNA Processing in Eukaryotes
After transcription, eukaryotic pre-mRNAs must undergo several processing steps before they can be translated. Eukaryotic (and prokaryotic) tRNAs and rRNAs also undergo processing before they can function as components in the protein synthesis machinery.
mRNA Processing
The eukaryotic pre-mRNA undergoes extensive processing before it is ready to be translated. The additional steps involved in eukaryotic mRNA maturation create a molecule with a much longer half-life than a prokaryotic mRNA. Eukaryotic mRNAs last for several hours, whereas the typical E. coli mRNA lasts no more than five seconds.
Pre-mRNAs are first coated in RNA-stabilizing proteins; these protect the pre-mRNA from degradation while it is processed and exported out of the nucleus. The three most important steps of pre-mRNA processing are the addition of stabilizing and signaling factors at the 5′ and 3′ ends of the molecule, and the removal of intervening sequences that do not specify the appropriate amino acids. In rare cases, the mRNA transcript can be “edited” after it is transcribed.
Pre-mRNA Splicing
Eukaryotic genes are composed of exons, which correspond to protein-coding sequences (ex-on signifies that they are expressed), and intervening sequences called introns (int-ron denotes their intervening role), which may be involved in gene regulation but are removed from the pre-mRNA during processing. Intron sequences in mRNA do not encode functional proteins.
The discovery of introns came as a surprise to researchers in the 1970s who expected that pre-mRNAs would specify protein sequences without further processing, as they had observed in prokaryotes. The genes of higher eukaryotes very often contain one or more introns. These regions may correspond to regulatory sequences; however, the biological significance of having many introns or having very long introns in a gene is unclear. It is possible that introns slow down gene expression because it takes longer to transcribe pre-mRNAs with lots of introns. Alternatively, introns may be nonfunctional sequence remnants left over from the fusion of ancient genes throughout evolution. This is supported by the fact that separate exons often encode separate protein subunits or domains. For the most part, the sequences of introns can be mutated without ultimately affecting the protein product.
All of a pre-mRNA’s introns must be completely and precisely removed before protein synthesis. If the process errs by even a single nucleotide, the reading frame of the rejoined exons would shift, and the resulting protein would be dysfunctional. The process of removing introns and reconnecting exons is called splicing (Figure 15.13). Introns are removed and degraded while the pre-mRNA is still in the nucleus. Splicing occurs by a sequence-specific mechanism that ensures introns will be removed and exons rejoined with the accuracy and precision of a single nucleotide. The splicing of pre-mRNAs is conducted by complexes of proteins and RNA molecules called spliceosomes.
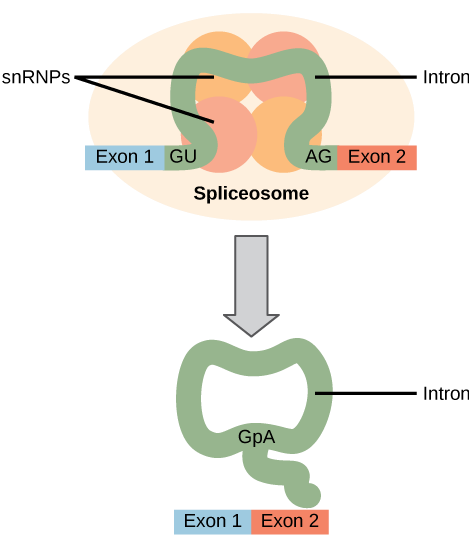
Note that more than 70 individual introns can be present, and each has to undergo the process of splicing—in addition to 5′ capping and the addition of a poly-A tail—just to generate a single, translatable mRNA molecule.
See how introns are removed during RNA splicing at this website (http://openstaxcollege.org/l/ RNA_splicing) .
Processing of tRNAs and rRNAs
The tRNAs and rRNAs are structural molecules that have roles in protein synthesis; however, these RNAs are not themselves translated. Pre-rRNAs are transcribed, processed, and assembled into ribosomes in the nucleolus. Pre-tRNAs are transcribed and processed in the nucleus and then released into the cytoplasm where they are linked to free amino acids for protein synthesis.
Most of the tRNAs and rRNAs in eukaryotes and prokaryotes are first transcribed as a long precursor molecule that spans multiple rRNAs or tRNAs. Enzymes then cleave the precursors into subunits corresponding to each structural RNA. Some of the bases of pre-rRNAs are methylated; that is, a –CH3 moiety (methyl functional group) is added for stability. Pre-tRNA molecules also undergo methylation. As with pre-mRNAs, subunit excision occurs in eukaryotic pre-RNAs destined to become tRNAs or rRNAs.
Mature rRNAs make up approximately 50 percent of each ribosome. Some of a ribosome’s RNA molecules are purely structural, whereas others have catalytic or binding activities. Mature tRNAs take on a three-dimensional structure through intramolecular hydrogen bonding to position the amino acid binding site at one end and the anticodon at the other end (Figure 15.14). The anticodon is a three nucleotide sequence in a tRNA that interacts with an mRNA codon through complementary base pairing.
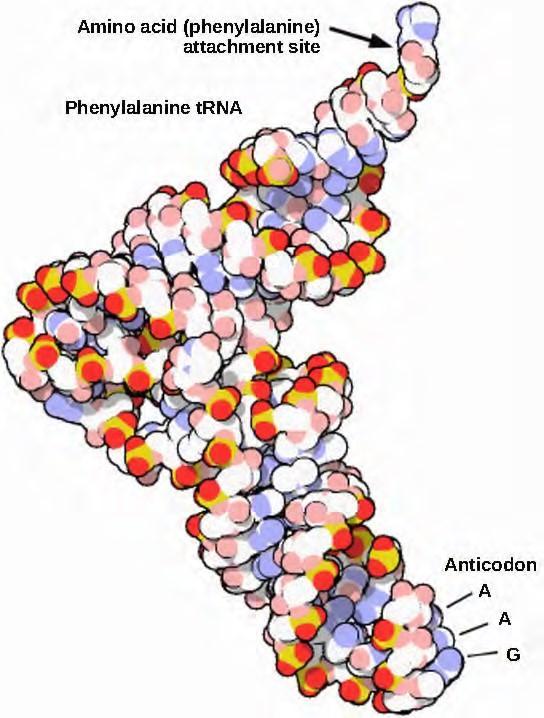
Figure 9.29 This is a space-filling model of a tRNA molecule that adds the amino acid phenylalanine to a growing polypeptide chain. The anticodon AAG binds the Codon UUC on the mRNA. The amino acid phenylalanine is attached to the other end of the tRNA.
9.10 | Ribosomes and Protein Synthesis
The synthesis of proteins consumes more of a cell’s energy than any other metabolic process. In turn, proteins account for more mass than any other component of living organisms (with the exception of water), and proteins perform virtually every function of a cell. The process of translation, or protein synthesis, involves the decoding of an mRNA message into a polypeptide product. Amino acids are covalently strung together by interlinking peptide bonds in lengths ranging from approximately 50 amino acid residues to more than 1,000. Each individual amino acid has an amino group (NH2) and a carboxyl (COOH) group. Polypeptides are formed when the amino group of one amino acid forms an amide (i.e., peptide) bond with the carboxyl group of another amino acid (Figure 9.30). This reaction is catalyzed by ribosomes and generates one water molecule.
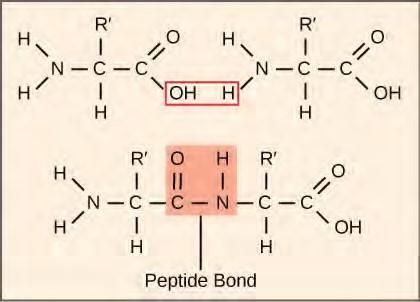
Figure 9.30 A peptide bond links the carboxyl end of one amino acid with the amino end of another, expelling one water molecule. For simplicity in this image, only the functional groups involved in the peptide bond are shown. The R and R’ designations refer to the rest of each amino acid structure.
The Protein Synthesis Machinery
In addition to the mRNA template, many molecules and macromolecules contribute to the process of translation. The composition of each component may vary across species; for instance, ribosomes may consist of different numbers of rRNAs and polypeptides depending on the organism. However, the general structures and functions of the protein synthesis machinery are comparable from bacteria to human cells. Translation requires the input of an mRNA template, ribosomes, tRNAs, and various enzymatic factors.
Click through the steps of this PBS interactive (http://openstaxcollege.org/l/prokary_protein) to see protein synthesis in action.
Ribosomes
Even before an mRNA is translated, a cell must invest energy to build each of its ribosomes. In E. coli, there are between 10,000 and 70,000 ribosomes present in each cell at any given time. A ribosome is a complex macromolecule composed of structural and catalytic rRNAs, and many distinct polypeptides. In eukaryotes, the nucleolus is completely specialized for the synthesis and assembly of rRNAs.
Ribosomes exist in the cytoplasm in prokaryotes and in the cytoplasm and rough endoplasmic reticulum in eukaryotes. Mitochondria and chloroplasts also have their own ribosomes in the matrix and stroma, which look more similar to prokaryotic ribosomes (and have similar drug sensitivities) than the ribosomes just outside their outer membranes in the cytoplasm. Ribosomes dissociate into large and small subunits when they are not synthesizing proteins and reassociate during the initiation of translation. In E. coli, the small subunit is described as 30S, and the large subunit is 50S, for a total of 70S (recall that Svedberg units are not additive). Mammalian ribosomes have a small 40S subunit and a large 60S subunit, for a total of 80S. The small subunit is responsible for binding the mRNA template, whereas the large subunit sequentially binds tRNAs. Each mRNA molecule is simultaneously translated by many ribosomes, all synthesizing protein in the same direction: reading the mRNA from 5′ to 3′ and synthesizing the polypeptide from the N terminus to the C terminus. The complete mRNA/poly-ribosome structure is called a polysome.
tRNAs
The tRNAs are structural RNA molecules that were transcribed from genes by RNA polymerase III. Depending on the species, 40 to 60 types of tRNAs exist in the cytoplasm. Serving as adaptors, specific tRNAs bind to sequences on the mRNA template and add the corresponding amino acid to the polypeptide chain. Therefore, tRNAs are the molecules that actually “translate” the language of RNA into the language of proteins.
Of the 64 possible mRNA codons—or triplet combinations of A, U, G, and C—three specify the termination of protein synthesis and 61 specify the addition of amino acids to the polypeptide chain. Of these 61, one codon (AUG) also encodes the initiation of translation. Each tRNA anticodon can base pair with one of the mRNA codons and add an amino acid or terminate translation, according to the genetic code. For instance, if the sequence CUA occurred on an mRNA template in the proper reading frame, it would bind a tRNA expressing the complementary sequence, GAU, which would be linked to the amino acid leucine.
As the adaptor molecules of translation, it is surprising that tRNAs can fit so much specificity into such a small package. Consider that tRNAs need to interact with three factors: 1) they must be recognized by the correct aminoacyl synthetase (see below); 2) they must be recognized by ribosomes; and 3) they must bind to the correct sequence in mRNA.
Aminoacyl tRNA Synthetases
The process of pre-tRNA synthesis by RNA polymerase III only creates the RNA portion of the adaptor molecule. The corresponding amino acid must be added later, once the tRNA is processed and exported to the cytoplasm. Through the process of tRNA “charging,” each tRNA molecule is linked to its correct amino acid by a group of enzymes called aminoacyl tRNA synthetases. At least one type of aminoacyl tRNA synthetase exists for each of the 20 amino acids; the exact number of aminoacyl tRNA synthetases varies by species. These enzymes first bind and hydrolyze ATP to catalyze a high-energy bond between an amino acid and adenosine monophosphate (AMP); a pyrophosphate molecule is expelled in this reaction. The activated amino acid is then transferred to the tRNA, and AMP is released.
The Mechanism of Protein Synthesis
As with mRNA synthesis, protein synthesis can be divided into three phases: initiation, elongation, and termination. The process of translation is similar in prokaryotes and eukaryotes. Here we’ll explore how translation occurs in E. coli, a representative prokaryote, and specify any differences between prokaryotic and eukaryotic translation.
Initiation of Translation
Protein synthesis begins with the formation of an initiation complex. In E. coli, this complex involves the small 30S ribosome, the mRNA template, three initiation factors (IFs; IF-1, IF-2, and IF-3), and a special initiator tRNA, called tRNAMetf . The initiator tRNA interacts with the start codon AUG (or rarely, GUG), links to a formylated methionine called fMet, and can also bind IF-2. Formylated methionine is inserted by fMet − tRNAMef t at the beginning of every polypeptide chain synthesized by E. coli, but it is usually clipped off after translation is complete. When an in-frame AUG is encountered during translation elongation, a non-formylated methionine is inserted by a regular Met-tRNAMet.
In E. coli mRNA, a sequence upstream of the first AUG codon, called the Shine-Dalgarno sequence (AGGAGG), interacts with the rRNA molecules that compose the ribosome. This interaction anchors the 30S ribosomal subunit at the correct location on the mRNA template. Guanosine triphosphate (GTP), which is a purine nucleotide triphosphate, acts as an energy source during translation—both at the start of elongation and during the ribosome’s translocation.
In eukaryotes, a similar initiation complex forms, comprising mRNA, the 40S small ribosomal subunit, IFs, and nucleoside triphosphates (GTP and ATP). The charged initiator tRNA, called Met-tRNAi, does not bind fMet in eukaryotes, but is distinct from other Met-tRNAs in that it can bind IFs.
Instead of depositing at the Shine-Dalgarno sequence, the eukaryotic initiation complex recognizes the
7-methylguanosine cap at the 5′ end of the mRNA. A cap-binding protein (CBP) and several other IFs assist the movement of the ribosome to the 5′ cap. Once at the cap, the initiation complex tracks along the mRNA in the 5′ to 3′ direction, searching for the AUG start codon. Many eukaryotic mRNAs are translated from the first AUG, but this is not always the case. According to Kozak’s rules, the nucleotides around the AUG indicate whether it is the correct start codon. Kozak’s rules state that the following consensus sequence must appear around the AUG of vertebrate genes: 5′-gccRccAUGG-3′. The R (for purine) indicates a site that can be either A or G, but cannot be C or U. Essentially, the closer the sequence is to this consensus, the higher the efficiency of translation.
Once the appropriate AUG is identified, the other proteins and CBP dissociate, and the 60S subunit binds to the complex of Met-tRNAi, mRNA, and the 40S subunit. This step completes the initiation of translation in eukaryotes.
Translation, Elongation, and Termination
In prokaryotes and eukaryotes, the basics of elongation are the same, so we will review elongation from the perspective of E. coli. The 50S ribosomal subunit of E. coli consists of three compartments: the A (aminoacyl) site binds incoming charged aminoacyl tRNAs. The P (peptidyl) site binds charged tRNAs carrying amino acids that have formed peptide bonds with the growing polypeptide chain but have not yet dissociated from their corresponding tRNA. The E (exit) site releases dissociated tRNAs so that they can be recharged with free amino acids. There is one exception to this assembly line of tRNAs: in E. coli, fMet − tRNAMetf is capable of entering the P site directly without first entering the A site. Similarly, the eukaryotic Met-tRNAi, with help from other proteins of the initiation complex, binds directly to the P site. In both cases, this creates an initiation complex with a free A site ready to accept the tRNA corresponding to the first codon after the AUG.
During translation elongation, the mRNA template provides specificity. As the ribosome moves along the mRNA, each mRNA codon comes into register, and specific binding with the corresponding charged tRNA anticodon is ensured. If mRNA were not present in the elongation complex, the ribosome would bind tRNAs nonspecifically.
Elongation proceeds with charged tRNAs entering the A site and then shifting to the P site followed by the E site with each single-codon “step” of the ribosome. Ribosomal steps are induced by conformational changes that advance the ribosome by three bases in the 3′ direction. The energy for each step of the ribosome is donated by an elongation factor that hydrolyzes GTP. Peptide bonds form between the amino group of the amino acid attached to the A-site tRNA and the carboxyl group of the amino acid attached to the P-site tRNA. The formation of each peptide bond is catalyzed by peptidyl transferase, an RNA-based enzyme that is integrated into the 50S ribosomal subunit. The energy for each peptide bond formation is derived from GTP hydrolysis, which is catalyzed by a separate elongation factor. The amino acid bound to the P-site tRNA is also linked to the growing polypeptide chain. As the ribosome steps across the mRNA, the former P-site tRNA enters the E site, detaches from the amino acid, and is expelled (Figure 14.31). Amazingly, the E. coli translation apparatus takes only 0.05 seconds to add each amino acid, meaning that a 200-amino acid protein can be translated in just 10 seconds.
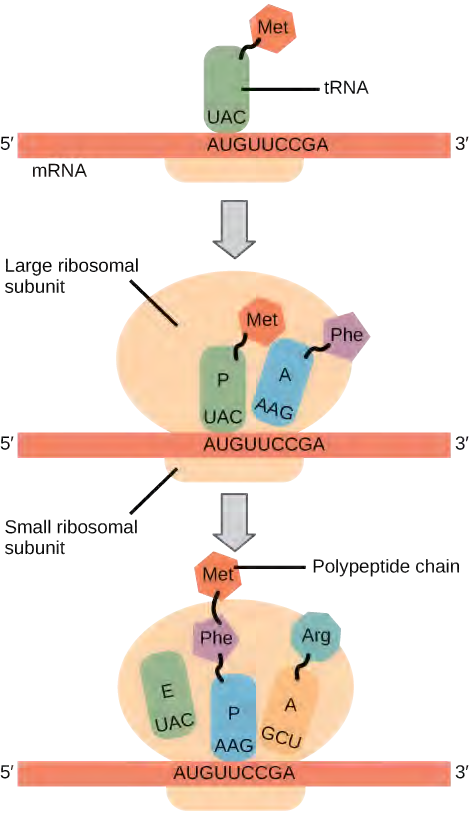
Termination of translation occurs when a nonsense codon (UAA, UAG, or UGA) is encountered. Upon aligning with the A site, these nonsense codons are recognized by release factors in prokaryotes and eukaryotes that instruct peptidyl transferase to add a water molecule to the carboxyl end of the P-site amino acid. This reaction forces the P-site amino acid to detach from its tRNA, and the newly made protein is released. The small and large ribosomal subunits dissociate from the mRNA and from each other; they are recruited almost immediately into another translation initiation complex. After many ribosomes have completed translation, the mRNA is degraded so the nucleotides can be reused in another transcription reaction.
Protein Folding, Modification, and Targeting
During and after translation, individual amino acids may be chemically modified, signal sequences may be appended, and the new protein “folds” into a distinct three-dimensional structure as a result of intramolecular interactions. A signal sequence is a short tail of amino acids that directs a protein to a specific cellular compartment. These sequences at the amino end or the carboxyl end of the protein can be thought of as the protein’s “train ticket” to its ultimate destination. Other cellular factors recognize each signal sequence and help transport the protein from the cytoplasm to its correct compartment. For instance, a specific sequence at the amino terminus will direct a protein to the mitochondria or chloroplasts (in plants). Once the protein reaches its cellular destination, the signal sequence is usually clipped off.
Many proteins fold spontaneously, but some proteins require helper molecules, called chaperones, to prevent them from aggregating during the complicated process of folding. Even if a protein is properly specified by its corresponding mRNA, it could take on a completely dysfunctional shape if abnormal temperature or pH conditions prevent it from folding correctly.
9.11 | Regulation of Gene Expression
For a cell to function properly, necessary proteins must be synthesized at the proper time. All cells control or regulate the synthesis of proteins from information encoded in their DNA. The process of turning on a gene to produce RNA and protein is called gene expression. Whether in a simple unicellular organism or a complex multi-cellular organism, each cell controls when and how its genes are expressed. For this to occur, there must be a mechanism to control when a gene is expressed to make RNA and protein, how much of the protein is made, and when it is time to stop making that protein because it is no longer needed.
Prokaryotic versus Eukaryotic Gene Expression
To understand how gene expression is regulated, we must first understand how a gene codes for a functional protein in a cell. The process occurs in both prokaryotic and eukaryotic cells, just in slightly different manners.Prokaryotic organisms are single-celled organisms that lack a cell nucleus, and their DNA therefore floats freely in the cell cytoplasm. To synthesize a protein, the processes of transcription and translation occur almost simultaneously. When the resulting protein is no longer needed, transcription stops. As a result, the primary method to control what type of protein and how much of each protein is expressed in a prokaryotic cell is the regulation of DNA transcription. All of the subsequent steps occur automatically. When more protein is required, more transcription occurs. Therefore, in prokaryotic cells, the control of gene expression is mostly at the transcriptional level.Eukaryotic cells, in contrast, have intracellular organelles that add to their complexity. In eukaryotic cells, the DNA is contained inside the cell’s nucleus and there it is transcribed into RNA. The newly synthesized RNA is then transported out of the nucleus into the cytoplasm, where ribosomes translate the RNA into protein. The processes of transcription and translation are physically separated by the nuclear membrane; transcription occurs only within the nucleus, and translation occurs only outside the nucleus in the cytoplasm. The regulation of gene expression can occur at all stages of the process (Figure 16.2). Regulation may occur when the DNA is uncoiled and loosened from nucleosomes to bind transcription factors ( epigenetic level), when the RNA is transcribed (transcriptional level), when the RNA is processed and exported to the cytoplasm after it is transcribed ( post-transcriptional level), when the RNA is translated into protein (translational level), or after the protein has been made ( post-translational level).
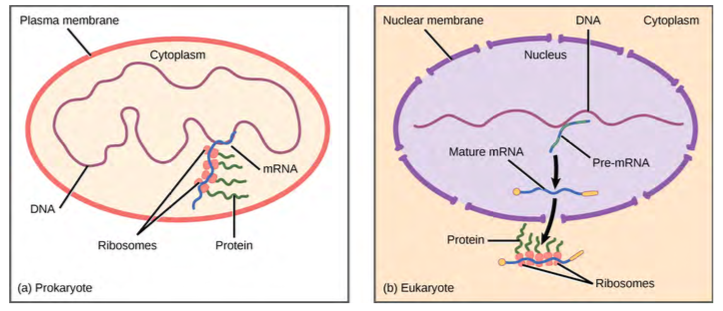
Figure 9.32 Prokaryotic transcription and translation occur simultaneously in the cytoplasm, and regulation occurs at the transcriptional level. Eukaryotic gene expression is regulated during transcription and RNA processing, which take place in the nucleus, and during protein translation, which takes place in the cytoplasm. Further regulation may occur through post-translational modifications of proteins.


9.12 | Prokaryotic Gene Regulation
The DNA of prokaryotes is organized into a circular chromosome supercoiled in the nucleoid region of the cell cytoplasm. Proteins that are needed for a specific function, or that are involved in the same biochemical pathway, are encoded together in blocks called operons. For example, all of the genes needed to use lactose as an energy source are coded next to each other in the lactose (or lac) operon.
In prokaryotic cells, there are three types of regulatory molecules that can affect the expression of operons: repressors, activators, and inducers. Repressors are proteins that suppress transcription of a gene in response to an external stimulus, whereas activators are proteins that increase the transcription of a gene in response to an external stimulus. Finally, inducers are small molecules that either activate or repress transcription depending on the needs of the cell and the availability of substrate.
The trp Operon: A Repressor Operon
Bacteria such as E. coli need amino acids to survive. Tryptophan is one such amino acid that E. coli can ingest from the environment. E. coli can also synthesize tryptophan using enzymes that are encoded by five genes. These five genes are next to each other in what is called the tryptophan (trp) operon (Figure 9.33). If tryptophan is present in the environment, then E. coli does not need to synthesize it and the switch controlling the activation of the genes in the trp operon is switched off. However, when tryptophan availability is low, the switch controlling the operon is turned on, transcription is initiated, the genes are expressed, and tryptophan is synthesized.
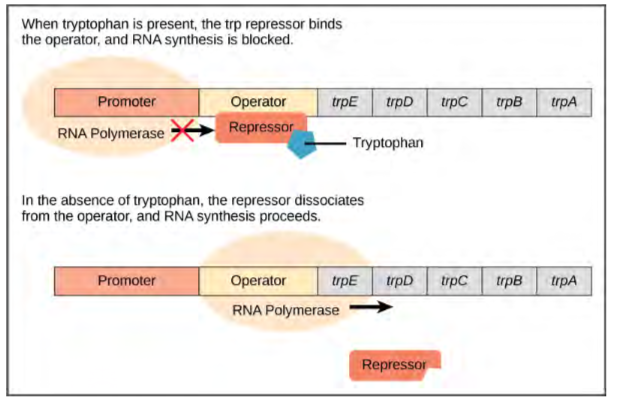
Figure 9.33 The five genes that are needed to synthesize tryptophan in E. coli are located next to each other in the trp operon. When tryptophan is plentiful, two tryptophan molecules bind the repressor protein at the operator sequence. This physically blocks the RNA polymerase from transcribing the tryptophan genes. When tryptophan is absent, the repressor protein does not bind to the operator and the genes are transcribed.A DNA sequence that codes for proteins is referred to as the coding region. The five coding regions for the tryptophan biosynthesis enzymes are arranged sequentially on the chromosome in the operon. Just before the coding region is the transcriptional start site. This is the region of DNA to which RNA polymerase binds to initiate transcription. The promoter sequence is upstream of the transcriptional start site; each operon has a sequence within or near the promoter to which proteins (activators or repressors) can bind and regulate transcription.
Watch this video (http://openstaxcollege.org/l/trp_operon) to learn more about the trp operon.
Catabolite Activator Protein (CAP): An Activator Regulator
Just as the trp operon is negatively regulated by tryptophan molecules, there are proteins that bind to the operator sequences that act as a positive regulator to turn genes on and activate them. For example, when glucose is scarce, E. coli bacteria can turn to other sugar sources for fuel. To do this, new genes to process these alternate genes must be transcribed. When glucose levels drop, cyclic AMP (cAMP) begins to accumulate in the cell. The cAMP molecule is a signaling molecule that is involved in glucose and energy metabolism in E. coli. When glucose levels decline in the cell, accumulating cAMP binds to the positive regulator catabolite activator protein (CAP), a protein that binds to the promoters of operons that control the processing of alternative sugars. When cAMP binds to CAP, the complex binds to the promoter region of the genes that are needed to use the alternate sugar sources (Figure 16.4). In these operons, a CAP binding site is located upstream of the RNA polymerase binding site in the promoter. This increases the binding ability of RNA polymerase to the promoter region and the transcription of the genes.
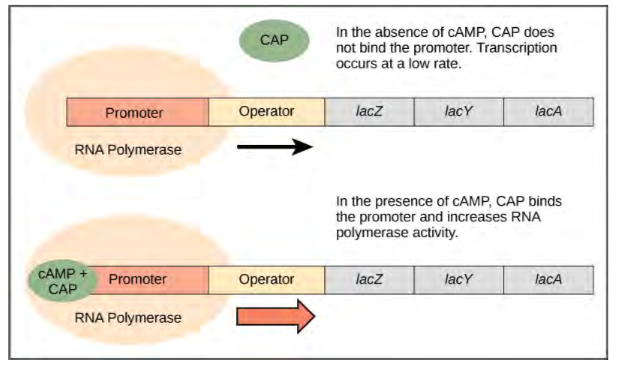
Figure 9.34 When glucose levels fall, E. coli may use other sugars for fuel but must transcribe new genes to do so. As glucose supplies become limited, cAMP levels increase. This cAMP binds to the CAP protein, a positive regulator that binds to an operator region upstream of the genes required to use other sugar sources.
The lac Operon: An Inducer Operon
The third type of gene regulation in prokaryotic cells occurs through inducible operons, which have proteins that bind to activate or repress transcription depending on the local environment and the needs of the cell. The lac operon is a typical inducible operon. As mentioned previously, E. coli is able to use other sugars as energy sources when glucose concentrations are low. To do so, the cAMP–CAP protein complex serves as a positive regulator to induce transcription. One such sugar source is lactose. The lac operon encodes the genes necessary to acquire and process the lactose from the local environment. CAP binds to the operator sequence upstream of the promoter that initiates transcription of the lac operon. However, for the lac operon to be activated, two conditions must be met. First, the level of glucose must be very low or non-existent. Second, lactose must be present. Only when glucose is absent and lactose is present will the lac operon be transcribed (Figure 16.5). This makes sense for the cell, because it would be energetically wasteful to create the proteins to process lactose if glucose was plentiful or lactose was not available.
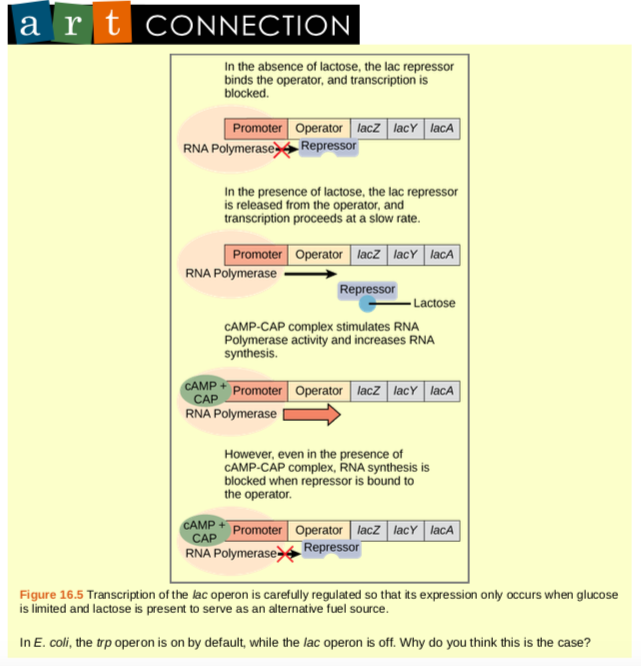
If glucose is absent, then CAP can bind to the operator sequence to activate transcription. If lactose is absent, then the repressor binds to the operator to prevent transcription. If either of these requirements is met, then transcription remains off. Only when both conditions are satisfied is the lac operon transcribed (Table 16.2).
Watch an animated tutorial (http://openstaxcollege.org/l/lac_operon) about the workings of lac operon here.
9.13 | Eukaryotic Epigenetic Gene Regulation
Eukaryotic gene expression is more complex than prokaryotic gene expression because the processes of transcription and translation are physically separated. Unlike prokaryotic cells, eukaryotic cells can regulate gene expression at many different levels. Eukaryotic gene expression begins with control of access to the DNA. This form of regulation, called epigenetic regulation, occurs even before transcription is initiated.
The first level of organization, or packing, is the winding of DNA strands around histone proteins. Histones package and order DNA into structural units called nucleosome complexes, which can control the access of proteins to the DNA regions (Figure 16.6a). Under the electron microscope, this winding of DNA around histone proteins to form nucleosomes looks like small beads on a string (Figure 16.6b). These beads (histone proteins) can move along the string (DNA) and change the structure of the molecule.
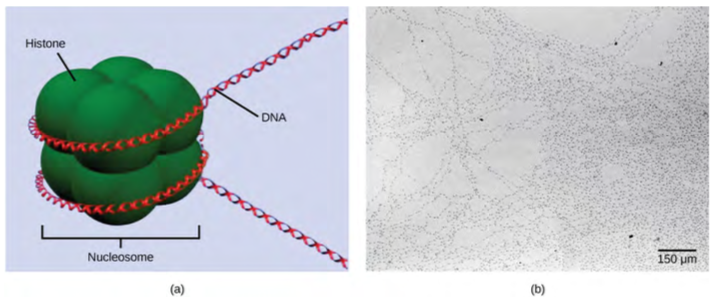
Figure 9.35 DNA is folded around histone proteins to create (a) nucleosome complexes. These nucleosomes control the access of proteins to the underlying DNA. When viewed through an electron microscope (b), the nucleosomes look like beads on a string. (credit “micrograph”: modification of work by Chris Woodcock)
If DNA encoding a specific gene is to be transcribed into RNA, the nucleosomes surrounding that region of DNA can slide down the DNA to open that specific chromosomal region and allow for the transcriptional machinery (RNA polymerase) to initiate transcription (Figure 9.35). Nucleosomes can move to open the chromosome structure to expose a segment of DNA, but do so in a very controlled manner.
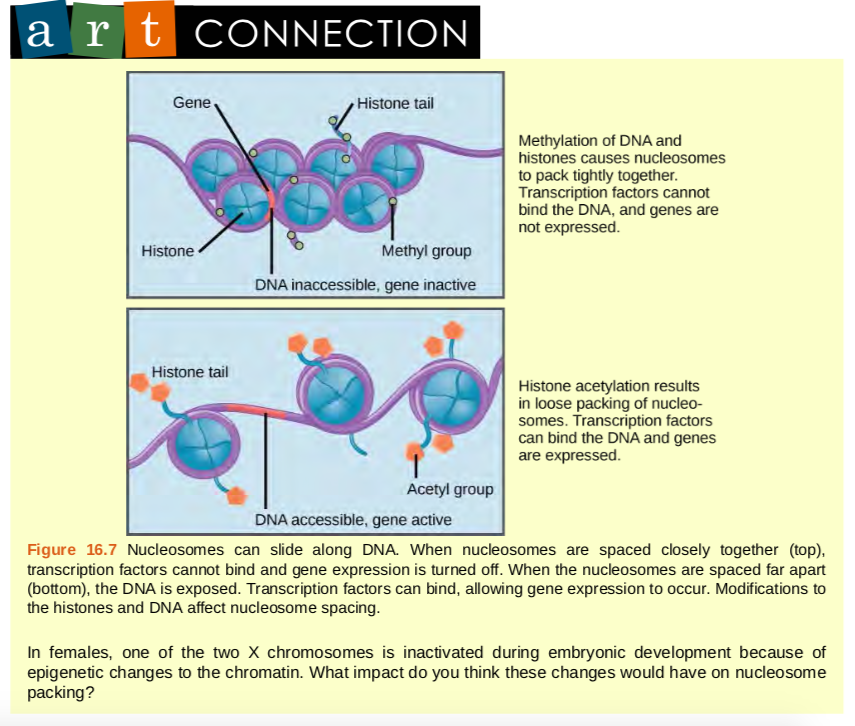
Figure 9.36 Nucleosomes can slide along DNA. When nucleosomes are spaced closely together (top), transcription factors cannot bind and gene expression is turned off. When the nucleosomes are spaced far apart (bottom), the DNA is exposed. Transcription factors can bind, allowing gene expression to occur. Modifications to the histones and DNA affect nucleosome spacing.
In females, one of the two X chromosomes is inactivated during embryonic development because of epigenetic changes to the chromatin. What impact do you think these changes would have on nucleosome packing?
How the histone proteins move is dependent on signals found on both the histone proteins and on the DNA. These signals are tags added to histone proteins and DNA that tell the histones if a chromosomal region should be open or closed (Figure 16.8 depicts modifications to histone proteins and DNA). These tags are not permanent, but may be added or removed as needed. They are chemical modifications (phosphate, methyl, or acetyl groups) that are attached to specific amino acids in the protein or to the nucleotides of the DNA. The tags do not alter the DNA base sequence, but they do alter how tightly wound the DNA is around the histone proteins. DNA is a negatively charged molecule; therefore, changes in the charge of the histone will change how tightly wound the DNA molecule will be. When unmodified, the histone proteins have a large positive charge; by adding chemical modifications like acetyl groups, the charge becomes less positive.
The DNA molecule itself can also be modified. This occurs within very specific regions called CpG islands. These are stretches with a high frequency of cytosine and guanine dinucleotide DNA pairs (CG) found in the promoter regions of genes. When this configuration exists, the cytosine member of the pair can be methylated (a methyl group is added). This modification changes how the DNA interacts with proteins, including the histone proteins that control access to the region. Highly methylated (hypermethylated) DNA regions with deacetylated histones are tightly coiled and transcriptionally inactive.
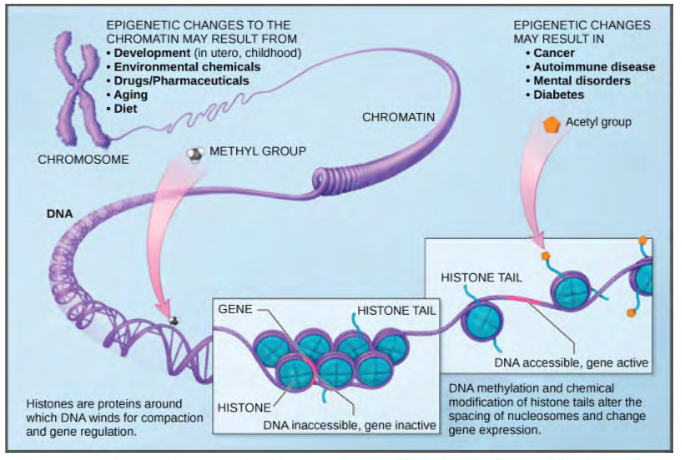
Figure 9.37 Histone proteins and DNA nucleotides can be modified chemically. Modifications affect nucleosome spacing and gene expression. (credit: modification of work by NIH)
View this video (http://openstaxcollege.org/l/epigenetic_reg) that describes how epigenetic regulation controls gene expression.
9.14 | Eukaryotic Transcription Gene Regulation
Like prokaryotic cells, the transcription of genes in eukaryotes requires the actions of an RNA polymerase to bind to a sequence upstream of a gene to initiate transcription. However, unlike prokaryotic cells, the eukaryotic RNA polymerase requires other proteins, or transcription factors, to facilitate transcription initiation. Transcription factors are proteins that bind to the promoter sequence and other regulatory sequences to control the transcription of the target gene. RNA polymerase by itself cannot initiate transcription in eukaryotic cells. Transcription factors must bind to the promoter region first and recruit RNA polymerase to the site for transcription to be established.
View the process of transcription—the making of RNA from a DNA template—at this site (http://openstaxcollege.org/ l/transcript_RNA) .
The Promoter and the Transcription Machinery
Genes are organized to make the control of gene expression easier. The promoter region is immediately upstream of the coding sequence. This region can be short (only a few nucleotides in length) or quite long (hundreds of nucleotides long). The longer the promoter, the more available space for proteins to bind. This also adds more control to the transcription process. The length of the promoter is gene-specific and can differ dramatically between genes. Consequently, the level of control of gene expression can also differ quite dramatically between genes. The purpose of the promoter is to bind transcription factors that control the initiation of transcription.
Within the promoter region, just upstream of the transcriptional start site, resides the TATA box. This box is simply a repeat of thymine and adenine dinucleotides (literally, TATA repeats). RNA polymerase binds to the transcription initiation complex, allowing transcription to occur. To initiate transcription, a transcription factor (TFIID) is the first to bind to the TATA box. Binding of TFIID recruits other transcription factors, including TFIIB, TFIIE, TFIIF, and TFIIH to the TATA box. Once this complex is assembled, RNA polymerase can bind to its upstream sequence. When bound along with the transcription factors, RNA polymerase is phosphorylated. This releases part of the protein from the DNA to activate the transcription initiation complex and places RNA polymerase in the correct orientation to begin transcription; DNA-bending protein brings the enhancer, which can be quite a distance from the gene, in contact with transcription factors and mediator proteins (Figure 16.9).
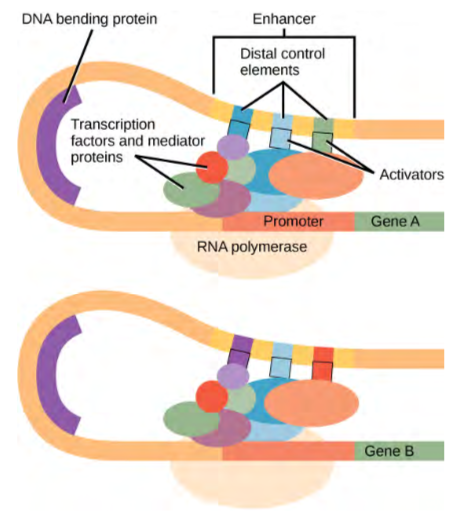
Figure 9.40 An enhancer is a DNA sequence that promotes transcription. Each enhancer is made up of short DNA sequences called distal control elements. Activators bound to the distal control elements interact with mediator proteins and transcription factors. Two different genes may have the same promoter but different distal control elements, enabling differential gene expression.
In addition to the general transcription factors, other transcription factors can bind to the promoter to regulate gene transcription. These transcription factors bind to the promoters of a specific set of genes. They are not general transcription factors that bind to every promoter complex, but are recruited to a specific sequence on the promoter of a specific gene. There are hundreds of transcription factors in a cell that each bind specifically to a particular DNA sequence motif. When transcription factors bind to the promoter just upstream of the encoded gene, it is referred to as a cis-acting element, because it is on the same chromosome just next to the gene. The region that a particular transcription factor binds to is called the transcription factor binding site. Transcription factors respond to environmental stimuli that cause the proteins to find their binding sites and initiate transcription of the gene that is needed.
Enhancers and Transcription
In some eukaryotic genes, there are regions that help increase or enhance transcription. These regions, called enhancers, are not necessarily close to the genes they enhance. They can be located upstream of a gene, within the coding region of the gene, downstream of a gene, or may be thousands of nucleotides away.
Enhancer regions are binding sequences, or sites, for transcription factors. When a DNA-bending protein binds, the shape of the DNA changes (Figure 16.9). This shape change allows for the interaction of the activators bound to the enhancers with the transcription factors bound to the promoter region and the RNA polymerase. Whereas DNA is generally depicted as a straight line in two dimensions, it is actually a three-dimensional object. Therefore, a nucleotide sequence thousands of nucleotides away can fold over and interact with a specific promoter.
Turning Genes Off: Transcriptional Repressors
Like prokaryotic cells, eukaryotic cells also have mechanisms to prevent transcription. Transcriptional repressors can bind to promoter or enhancer regions and block transcription. Like the transcriptional activators, repressors respond to external stimuli to prevent the binding of activating transcription factors.
9.15 | Eukaryotic Post-transcriptional Gene Regulation
RNA is transcribed, but must be processed into a mature form before translation can begin. This processing after an RNA molecule has been transcribed, but before it is translated into a protein, is called post-transcriptional modification. As with the epigenetic and transcriptional stages of processing, this post-transcriptional step can also be regulated to control gene expression in the cell. If the RNA is not processed, shuttled, or translated, then no protein will be synthesized.
RNA splicing, the first stage of post-transcriptional control
In eukaryotic cells, the RNA transcript often contains regions, called introns, that are removed prior to translation. The regions of RNA that code for protein are called exons (Figure 16.10). After an RNA molecule has been transcribed, but prior to its departure from the nucleus to be translated, the RNA is processed and the introns are removed by splicing.
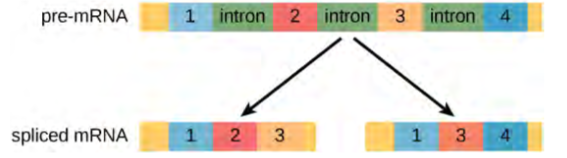
Figure 9.41 Pre-mRNA can be alternatively spliced to create different proteins.
Alternative RNA Splicing
In the 1970s, genes were first observed that exhibited alternative RNA splicing. Alternative RNA splicing is a mechanism that allows different protein products to be produced from one gene when different combinations of introns, and sometimes exons, are removed from the transcript. This alternative splicing can be haphazard, but more often it is controlled and acts as a mechanism of gene regulation, with the frequency of different splicing alternatives controlled by the cell as a way to control the production of different protein products in different cells or at different stages of development. Alternative splicing is now understood to be a common mechanism of gene regulation in eukaryotes; according to one estimate, 70 percent of genes in humans are expressed as multiple proteins through alternative splicing.
Control of RNA Stability
Binding of proteins to the RNA can influence its stability. Proteins, called RNA-binding proteins, or RBPs, can bind to the regions of the RNA just upstream or downstream of the protein-coding region. These regions in the RNA that are not translated into protein are called the untranslated regions, or UTRs. They are not introns (those have been removed in the nucleus). Rather, these are regions that regulate mRNA localization, stability, and protein translation. The region just before the protein-coding region is called the 5′ UTR, whereas the region after the coding region is called the 3′ UTR (Figure 16.12). The binding of RBPs to these regions can increase or decrease the stability of an RNA molecule, depending on the specific RBP that binds.
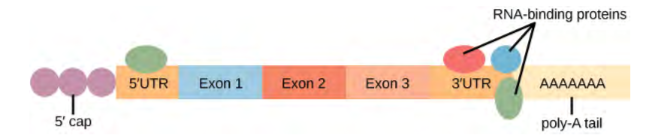
Figure 9.43 The protein-coding region of mRNA is flanked by 5′ and 3′ untranslated regions (UTRs). The presence of RNA-binding proteins at the 5′ or 3′ UTR influences the stability of the RNA molecule.
9.16 | Eukaryotic Translational and Post-translational Gene Regulation
After the RNA has been transported to the cytoplasm, it is translated into protein. Control of this process is largely dependent on the RNA molecule. As previously discussed, the stability of the RNA will have a large impact on its translation into a protein. As the stability changes, the amount of time that it is available for translation also changes.
The Initiation Complex and Translation Rate
Like transcription, translation is controlled by proteins that bind and initiate the process. In translation, the complex that assembles to start the process is referred to as the initiation complex. The first protein to bind to the RNA to initiate translation is the eukaryotic initiation factor-2 (eIF-2). The eIF-2 protein is active when it binds to the high-energy molecule guanosine triphosphate (GTP). GTP provides the energy to start the reaction by giving up a phosphate and becoming guanosine diphosphate (GDP). The eIF-2 protein bound to GTP binds to the small 40S ribosomal subunit. When bound, the methionine initiator tRNA associates with the eIF-2/40S ribosome complex, bringing along with it the mRNA to be translated. At this point, when the initiator complex is assembled, the GTP is converted into GDP and energy is released. The phosphate and the eIF-2 protein are released from the complex and the large 60S ribosomal subunit binds to translate the RNA. The binding of eIF-2 to the RNA is controlled by phosphorylation. If eIF-2 is phosphorylated, it undergoes a conformational change and cannot bind to GTP. Therefore, the initiation complex cannot form properly and translation is impeded (Figure 16.13). When eIF-2 remains unphosphorylated, it binds the RNA and actively translates the protein.
Chemical Modifications, Protein Activity, and Longevity
Proteins can be chemically modified with the addition of groups including methyl, phosphate, acetyl, and ubiquitin groups. The addition or removal of these groups from proteins regulates their activity or the length of time they exist in the cell.
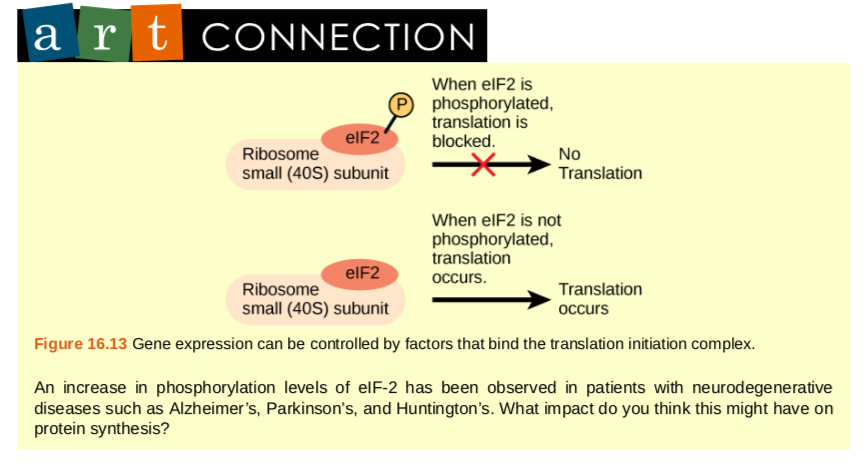
Figure 9.41 Gene expression can be controlled by factors that bind the translation initiation complex.An increase in phosphorylation levels of eIF-2 has been observed in patients with neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s. What impact do you think this might have on protein synthesis?Sometimes these modifications can regulate where a protein is found in the cell—for example, in the nucleus, the cytoplasm, or attached to the plasma membrane.
The addition of an ubiquitin group to a protein marks that protein for degradation. Ubiquitin acts like a flag indicating that the protein lifespan is complete. These proteins are moved to the proteasome, an organelle that functions to remove proteins, to be degraded (Figure 16.14). One way to control gene expression, therefore, is to alter the longevity of the protein.
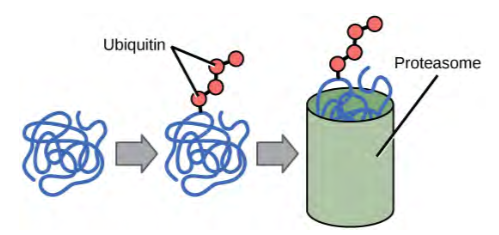
Figure 9.41 Proteins with ubiquitin tags are marked for degradation within the proteasome.
9.17 | Cancer and Gene Regulation
Cancer is not a single disease but includes many different diseases. In cancer cells, mutations modify cell-cycle control and cells don’t stop growing as they normally would. Mutations can also alter the growth rate or the progression of the cell through the cell cycle. One example of a gene modification that alters the growth rate is increased phosphorylation of cyclin B, a protein that controls the progression of a cell through the cell cycle and serves as a cell-cycle checkpoint protein.
For cells to move through each phase of the cell cycle, the cell must pass through checkpoints. This ensures that the cell has properly completed the step and has not encountered any mutation that will alter its function. Many proteins, including cyclin B, control these checkpoints. The phosphorylation of cyclin B, a post-translational event, alters its function. As a result, cells can progress through the cell cycle unimpeded, even if mutations exist in the cell and its growth should be terminated. This post-translational change of cyclin B prevents it from controlling the cell cycle and contributes to the development of cancer.
Cancer can be described as a disease of altered gene expression. There are many proteins that are turned on or off (gene activation or gene silencing) that dramatically alter the overall activity of the cell. A gene that is not normally expressed in that cell can be switched on and expressed at high levels. This can be the result of gene mutation or changes in gene regulation (epigenetic, transcription, post-transcription, translation, or post-translation).
Changes in epigenetic regulation, transcription, RNA stability, protein translation, and post-translational control can be detected in cancer. While these changes don’t occur simultaneously in one cancer, changes at each of these levels can be detected when observing cancer at different sites in different individuals. Therefore, changes in histone acetylation (epigenetic modification that leads to gene silencing), activation of transcription factors by phosphorylation, increased RNA stability, increased translational control, and protein modification can all be detected at some point in various cancer cells. Scientists are working to understand the common changes that give rise to certain types of cancer or how a modification might be exploited to destroy a tumor cell.
Tumor Suppressor Genes, Oncogenes, and Cancer
In normal cells, some genes function to prevent excess, inappropriate cell growth. These are tumor suppressor genes, which are active in normal cells to prevent uncontrolled cell growth. There are many tumor suppressor genes in cells. The most studied tumor suppressor gene is p53, which is mutated in over 50 percent of all cancer types. The p53 protein itself functions as a transcription factor. It can bind to sites in the promoters of genes to initiate transcription. Therefore, the mutation of p53 in cancer will dramatically alter the transcriptional activity of its target genes.
Watch this animation (http://openstaxcollege.org/l/p53_cancer) to learn more about the use of p53 in fighting cancer.
Proto-oncogenes are positive cell-cycle regulators. When mutated, proto-oncogenes can become oncogenes and cause cancer. Overexpression of the oncogene can lead to uncontrolled cell growth. This is because oncogenes can alter transcriptional activity, stability, or protein translation of another gene that directly or indirectly controls cell growth. An example of an oncogene involved in cancer is a protein called myc. Myc is a transcription factor that is aberrantly activated in Burkett’s Lymphoma, a cancer of the lymph system. Overexpression of myc transforms normal B cells into cancerous cells that continue to grow uncontrollably. High B-cell numbers can result in tumors that can interfere with normal bodily function. Patients with Burkett’s lymphoma can develop tumors on their jaw or in their mouth that interfere with the ability to eat.
Cancer and Epigenetic Alterations
Silencing genes through epigenetic mechanisms is also very common in cancer cells. There are characteristic modifications to histone proteins and DNA that are associated with silenced genes. In cancer cells, the DNA in the promoter region of silenced genes is methylated on cytosine DNA residues in CpG islands. Histone proteins that surround that region lack the acetylation modification that is present when the genes are expressed in normal cells. This combination of DNA methylation and histone deacetylation (epigenetic modifications that lead to gene silencing) is commonly found in cancer. When these modifications occur, the gene present in that chromosomal region is silenced. Increasingly, scientists understand how epigenetic changes are altered in cancer. Because these changes are temporary and can be reversed—for example, by preventing the action of the histone deacetylase protein that removes acetyl groups, or by DNA methyl transferase enzymes that add methyl groups to cytosines in DNA—it is possible to design new drugs and new therapies to take advantage of the reversible nature of these processes. Indeed, many researchers are testing how a silenced gene can be switched back on in a cancer cell to help re-establish normal growth patterns.
Genes involved in the development of many other illnesses, ranging from allergies to inflammation to autism, are thought to be regulated by epigenetic mechanisms. As our knowledge of how genes are controlled deepens, new ways to treat diseases like cancer will emerge.
Cancer and Transcriptional Control
Alterations in cells that give rise to cancer can affect the transcriptional control of gene expression. Mutations that activate transcription factors, such as increased phosphorylation, can increase the binding of a transcription factor to its binding site in a promoter. This could lead to increased transcriptional activation of that gene that results in modified cell growth. Alternatively, a mutation in the DNA of a promoter or enhancer region can increase the binding ability of a transcription factor. This could also lead to the increased transcription and aberrant gene expression that is seen in cancer cells.
Researchers have been investigating how to control the transcriptional activation of gene expression in cancer. Identifying how a transcription factor binds, or a pathway that activates where a gene can be turned off, has led to new drugs and new ways to treat cancer. In breast cancer, for example, many proteins are over expressed. This can lead to increased phosphorylation of key transcription factors that increase transcription. One such example is the over expression of the epidermal growth factor receptor (EGFR) in a subset of breast cancers. The EGFR pathway activates many protein kinases that, in turn, activate many transcription factors that control genes involved in cell growth. New drugs that prevent the activation of EGFR have been developed and are used to treat these cancers.
Cancer and Post-transcriptional Control
Changes in the post-transcriptional control of a gene can also result in cancer. Recently, several groups of researchers have shown that specific cancers have altered expression of miRNAs. Because miRNAs bind to the 3′ UTR of RNA molecules to degrade them, over expression of these miRNAs could be detrimental to normal cellular activity. Too many miRNAs could dramatically decrease the RNA population leading to a decrease in protein expression. Several studies have demonstrated a change in the miRNA population in specific cancer types. It appears that the subset of miRNAs expressed in breast cancer cells is quite different from the subset expressed in lung cancer cells or even from normal breast cells. This suggests that alterations in miRNA activity can contribute to the growth of breast cancer cells. These types of studies also suggest that if some miRNAs are specifically expressed only in cancer cells, they could be potential drug targets. It would, therefore, be conceivable that new drugs that turn off miRNA expression in cancer could be an effective method to treat cancer.
Cancer and Translational/Post-translational Control
There are many examples of how translational or post-translational modifications of proteins arise in cancer. Modifications are found in cancer cells from the increased translation of a protein to changes in protein phosphorylation to alternative splice variants of a protein. An example of how the expression of an alternative form of a protein can have dramatically different outcomes is seen in colon cancer cells. The c-Flip protein, a protein involved in mediating the cell death pathway, comes in two forms: long (c-FLIPL) and short (c-FLIPS). Both forms appear to be involved in initiating controlled cell death mechanisms in normal cells. However, in colon cancer cells, expression of the long form results in increased cell growth instead of cell death. Clearly, the expression of the wrong protein dramatically alters cell function and contributes to the development of cancer.
New Drugs to Combat Cancer: Targeted Therapies
Scientists are using what is known about the regulation of gene expression in disease states, including cancer, to develop new ways to treat and prevent disease development. Many scientists are designing drugs on the basis of the gene expression patterns within individual tumors. This idea, that therapy and medicines can be tailored to an individual, has given rise to the field of personalized medicine. With an increased understanding of gene regulation and gene function, medicines can be designed to specifically target diseased cells without harming healthy cells. Some new medicines, called targeted therapies, have exploited the overexpression of a specific protein or the mutation of a gene to develop a new medication to treat disease. One such example is the use of anti-EGF receptor medications to treat the subset of breast cancer tumors that have very high levels of the EGF protein. Undoubtedly, more targeted therapies will be developed as scientists learn more about how gene expression changes can cause cancer.
9.18 | Biotechnology
The study of nucleic acids began with the discovery of DNA, progressed to the study of genes and small fragments, and has now exploded to the field of genomics. Genomics is the study of entire genomes, including the complete set of genes, their nucleotide sequence and organization, and their interactions within a species and with other species. The advances in genomics have been made possible by DNA sequencing technology. Just as information technology has led to Google maps that enable people to get detailed information about locations around the globe, genomic information is used to create similar maps of the DNA of different organisms. These findings have helped anthropologists to better understand human migration and have aided the field of medicine through the mapping of human genetic diseases. The ways in which genomic information can contribute to scientific understanding are varied and quickly growing.
Biotechnology is the use of biological agents for technological advancement. Biotechnology was used for breeding livestock and crops long before the scientific basis of these techniques was understood. Since the discovery of the structure of DNA in 1953, the field of biotechnology has grown rapidly through both academic research and private companies. The primary applications of this technology are in medicine (production of vaccines and antibiotics) and agriculture (genetic modification of crops, such as to increase yields). Biotechnology also has many industrial applications, such as fermentation, the treatment of oil spills, and the production of biofuels (Figure 14.42).
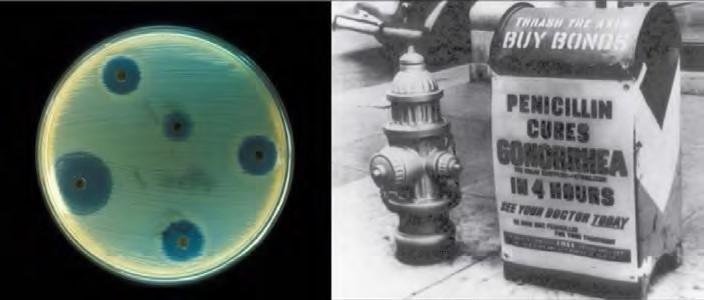
Figure 9.42 Antibiotics are chemicals produced by fungi, bacteria, and other organisms that have antimicrobial properties. The first antibiotic discovered was penicillin. Antibiotics are now commercially produced and tested for their potential to inhibit bacterial growth. (credit “advertisement”: modification of work by NIH; credit “test plate”: modification of work by Don Stalons/ CDC; scale-bar data from Matt Russell)
Basic Techniques to Manipulate Genetic Material (DNA and RNA)
To understand the basic techniques used to work with nucleic acids, remember that nucleic acids are macromolecules made of nucleotides (a sugar, a phosphate, and a nitrogenous base) linked by phosphodiester bonds. The phosphate groups on these molecules each have a net negative charge. An entire set of DNA molecules in the nucleus is called the genome. DNA has two complementary strands linked by hydrogen bonds between the paired bases. The two strands can be separated by exposure to high temperatures (DNA denaturation) and can be reannealed by cooling. The DNA can be replicated by the DNA polymerase enzyme. Unlike DNA, which is located in the nucleus of eukaryotic cells, RNA molecules leave the nucleus. The most common type of RNA that is analyzed is the messenger RNA (mRNA) because it represents the protein-coding genes that are actively expressed. However, RNA molecules present some other challenges to analysis, as they are often less stable than DNA.
DNA and RNA Extraction
To study or manipulate nucleic acids, the DNA or RNA must first be isolated or extracted from the cells. Various techniques are used to extract different types of DNA (Figure 9.43). Most nucleic acid extraction techniques involve steps to break open the cell and use enzymatic reactions to destroy all macromolecules that are not desired (such as degradation of unwanted molecules and separation from the DNA sample). Cells are broken using a lysis buffer (a solution which is mostly a detergent); lysis means “to split.” These enzymes break apart lipid molecules in the cell membranes and nuclear membranes. Macromolecules are inactivated using enzymes such as proteases that break down proteins, and ribonucleases (RNAses) that break down RNA. The DNA is then precipitated using alcohol. Human genomic DNA is usually visible as a gelatinous, white mass. The DNA samples can be stored frozen at –80°C for several years.
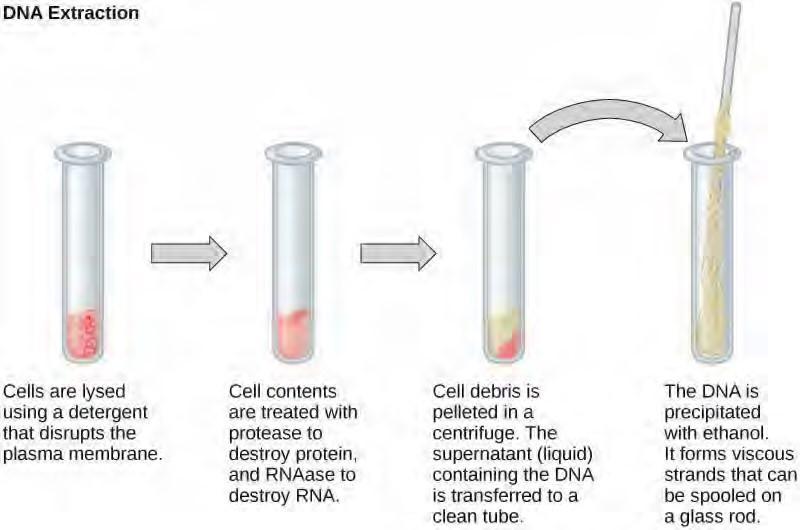
Figure 9.43 This diagram shows the basic method used for extraction of DNA.
RNA analysis is performed to study gene expression patterns in cells. RNA is naturally very unstable because RNAses are commonly present in nature and very difficult to inactivate. Similar to DNA, RNA extraction involves the use of various buffers and enzymes to inactivate macromolecules and preserve the RNA.
Gel Electrophoresis
Because nucleic acids are negatively charged ions at neutral or basic pH in an aqueous environment, they can be mobilized by an electric field. Gel electrophoresis is a technique used to separate molecules on the basis of size, using this charge. The nucleic acids can be separated as whole chromosomes or fragments. The nucleic acids are loaded into a slot near the negative electrode of a semisolid, porous gel matrix and pulled toward the positive electrode at the opposite end of the gel. Smaller molecules move through the pores in the gel faster than larger molecules; this difference in the rate of migration separates the fragments on the basis of size. There are molecular weight standard samples that can be run alongside the molecules to provide a size comparison. Nucleic acids in a gel matrix can be observed using various fluorescent or colored dyes. Distinct nucleic acid fragments appear as bands at specific distances from the top of the gel (the negative electrode end) on the basis of their size (Figure 9.44). A mixture of genomic DNA fragments of varying sizes appear as a long smear, whereas uncut genomicDNA is usually too large to run through the gel and forms a single large band at the top of the gel.
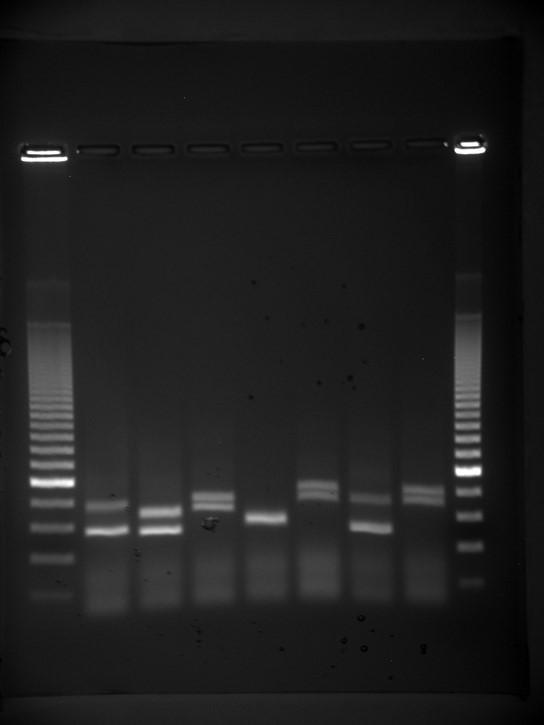
Figure 9.44 Shown are DNA fragments from seven samples run on a gel, stained with a fluorescent dye, and viewed under UV light. (credit: James Jacob, Tompkins Cortland Community College)
Amplification of Nucleic Acid Fragments by Polymerase Chain Reaction
Although genomic DNA is visible to the naked eye when it is extracted in bulk, DNA analysis often requires focusing on one or more specific regions of the genome. Polymerase chain reaction (PCR) is a technique used to amplify specific regions of DNA for further analysis (Figure 9.45). PCR is used for many purposes in laboratories, such as the cloning of gene fragments to analyze genetic diseases, identification of contaminant foreign DNA in a sample, and the amplification of DNA for sequencing.
More practical applications include the determination of paternity and detection of genetic diseases.
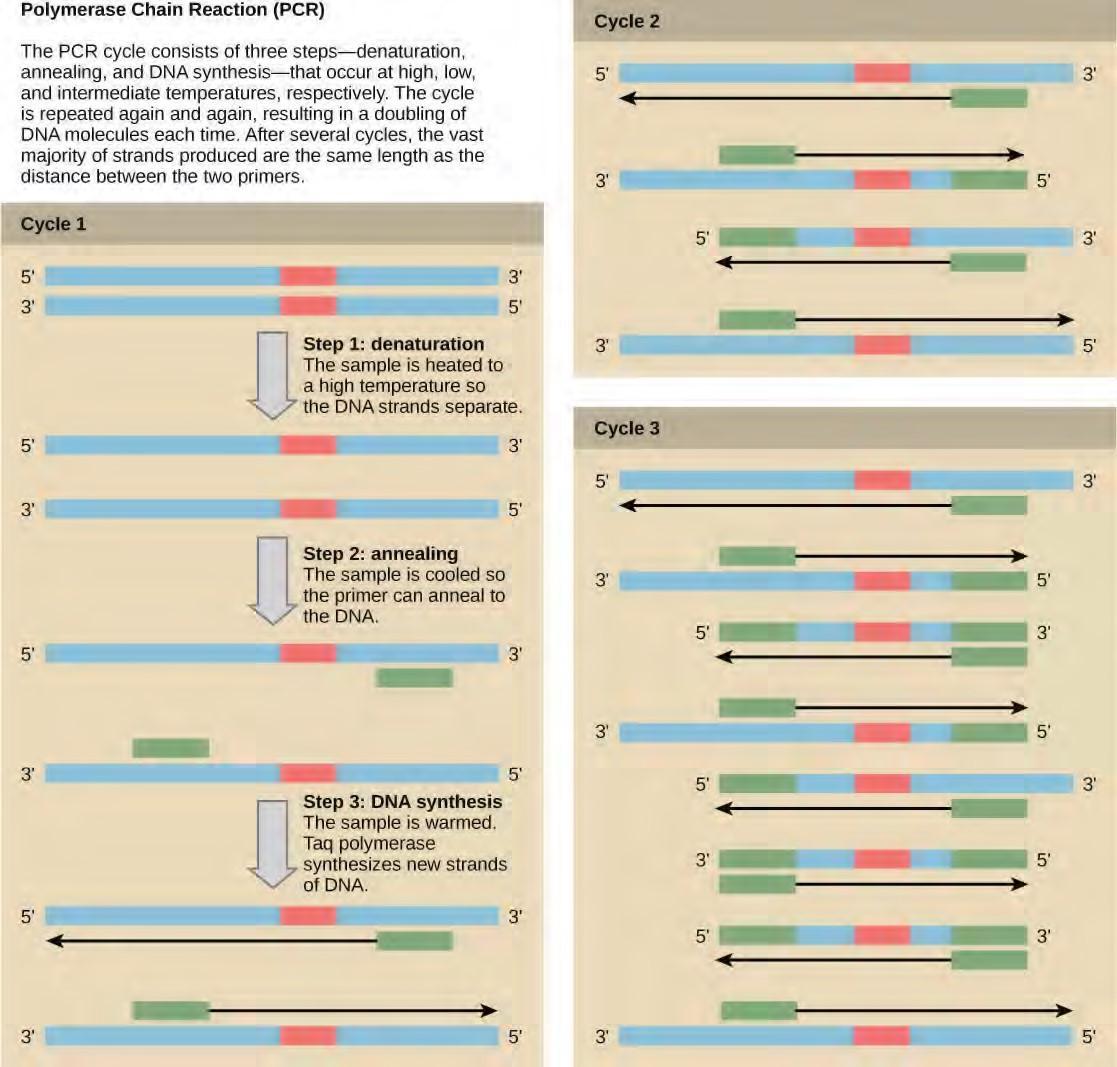
Figure 9.45 Polymerase chain reaction, or PCR, is used to amplify a specific sequence of DNA. Primers—short pieces of DNA complementary to each end of the target sequence—are combined with genomic DNA, Taq polymerase, and deoxynucleotides. Taq polymerase is a DNA polymerase isolated from the thermostable bacterium Thermus aquaticus that is able to withstand the high temperatures used in PCR. Thermus aquaticus grows in the Lower Geyser Basin of Yellowstone National Park. Reverse transcriptase PCR (RT-PCR) is similar to PCR, but cDNA is made from an RNA template before PCR begins.
DNA fragments can also be amplified from an RNA template in a process called reverse transcriptase PCR (RT-PCR). The first step is to recreate the original DNA template strand (called cDNA) by applying DNA nucleotides to the mRNA. This process is called reverse transcription. This requires the presence of an enzyme called reverse transcriptase. After the cDNA is made, regular PCR can be used to amplify it.
Deepen your understanding of the polymerase chain reaction by clicking through this interactive exercise (http://openstaxcollege.org/l/PCR) .
Molecular Cloning
In general, the word “cloning” means the creation of a perfect replica; however, in biology, the recreation of a whole organism is referred to as “reproductive cloning.” Long before attempts were made to clone an entire organism, researchers learned how to reproduce desired regions or fragments of the genome, a process that is referred to as molecular cloning.
Cloning small fragments of the genome allows for the manipulation and study of specific genes (and their protein products), or noncoding regions in isolation. A plasmid (also called a vector) is a small circular DNA molecule that replicates independently of the chromosomal DNA. In cloning, the plasmid molecules can be used to provide a “folder” in which to insert a desired DNA fragment. Plasmids are usually introduced into a bacterial host for proliferation. In the bacterial context, the fragment of DNA from the human genome (or the genome of another organism that is being studied) is referred to as foreign DNA, or a transgene, to differentiate it from the DNA of the bacterium, which is called the host DNA.
Plasmids occur naturally in bacterial populations (such as Escherichia coli) and have genes that can contribute favorable traits to the organism, such as antibiotic resistance (the ability to be unaffected by antibiotics). Plasmids have been repurposed and engineered as vectors for molecular cloning and the large-scale production of important reagents, such as insulin and human growth hormone. An important feature of plasmid vectors is the ease with which a foreign DNA fragment can be introduced via the multiple cloning site (MCS). The MCS is a short DNA sequence containing multiple sites that can be cut with different commonly available restriction endonucleases. Restriction endonucleases recognize specific DNA sequences and cut them in a predictable manner; they are naturally produced by bacteria as a defense mechanism against foreign DNA. Many restriction endonucleases make staggered cuts in the two strands of DNA, such that the cut ends have a 2- or 4-base single-stranded overhang. Because these overhangs are capable of annealing with complementary overhangs, these are called “sticky ends.” Addition of an enzyme called DNA ligase permanently joins the DNA fragments via phosphodiester bonds. In this way, any DNA fragment generated by restriction endonuclease cleavage can be spliced between the two ends of a plasmid DNA that has been cut with the same restriction endonuclease (Figure
9.47).
Recombinant DNA Molecules
Plasmids with foreign DNA inserted into them are called recombinant DNA molecules because they are created artificially and do not occur in nature. They are also called chimeric molecules because the origin of different parts of the molecules can be traced back to different species of biological organisms or even to chemical synthesis. Proteins that are expressed from recombinant DNA molecules are called recombinant proteins. Not all recombinant plasmids are capable of expressing genes. The recombinant DNA may need to be moved into a different vector (or host) that is better designed for gene expression. Plasmids may also be engineered to express proteins only when stimulated by certain environmental factors, so that scientists can control the expression of the recombinant proteins.

Figure 9.47. Cloning a gene in bacteria
View an animation of recombination in cloning (http://openstaxcollege.org/l/recombination) from the DNA Learning Center.
Cellular Cloning
Unicellular organisms, such as bacteria and yeast, naturally produce clones of themselves when they replicate asexually by binary fission; this is known as cellular cloning. The nuclear DNA duplicates by the process of mitosis, which creates an exact replica of the genetic material.
Reproductive Cloning
Reproductive cloning is a method used to make a clone or an identical copy of an entire multicellular organism. Most multicellular organisms undergo reproduction by sexual means, which involves genetic hybridization of two individuals (parents), making it impossible for generation of an identical copy or a clone of either parent. Recent advances in biotechnology have made it possible to artificially induce asexual reproduction of mammals in the laboratory.
Parthenogenesis, or “virgin birth,” occurs when an embryo grows and develops without the fertilization of the egg occurring; this is a form of asexual reproduction. An example of parthenogenesis occurs in species in which the female lays an egg and if the egg is fertilized, it is a diploid egg and the individual develops into a female; if the egg is not fertilized, it remains a haploid egg and develops into a male. The unfertilized egg is called a parthenogenic, or virgin, egg. Some insects and reptiles lay parthenogenic eggs that can develop into adults.
Sexual reproduction requires two cells; when the haploid egg and sperm cells fuse, a diploid zygote results. The zygote nucleus contains the genetic information to produce a new individual. However, early embryonic development requires the cytoplasmic material contained in the egg cell. This idea forms the basis for reproductive cloning. Therefore, if the haploid nucleus of an egg cell is replaced with a diploid nucleus from the cell of any individual of the same species (called a donor), it will become a zygote that is genetically identical to the donor. Somatic cell nuclear transfer is the technique of transferring a diploid nucleus into an enucleated egg. It can be used for either therapeutic cloning or reproductive cloning.
The first cloned animal was Dolly, a sheep who was born in 1996. The success rate of reproductive cloning at the time was very low. Dolly lived for seven years and died of respiratory complications (Figure 9.48). There is speculation that because the cell DNA belongs to an older individual, the age of the DNA may affect the life expectancy of a cloned individual. Since Dolly, several animals such as horses, bulls, and goats have been successfully cloned, although these individuals often exhibit facial, limb, and cardiac abnormalities. There have been attempts at producing cloned human embryos as sources of embryonic stem cells, sometimes referred to as cloning for therapeutic purposes. Therapeutic cloning produces stem cells to attempt to remedy detrimental diseases or defects (unlike reproductive cloning, which aims to reproduce an organism). Still, therapeutic cloning efforts have met with resistance because of bioethical considerations.
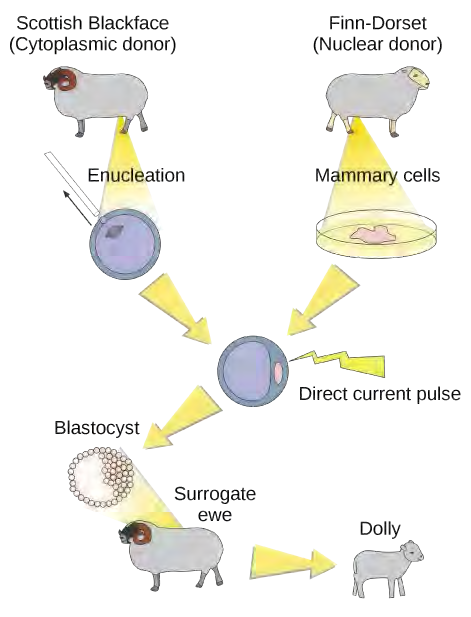
Genetic Engineering
Genetic engineering is the alteration of an organism’s genotype using recombinant DNA technology to modify an organism’s DNA to achieve desirable traits. The addition of foreign DNA in the form of recombinant DNA vectors generated by molecular cloning is the most common method of genetic engineering. The organism that receives the recombinant DNA is called a genetically modified organism (GMO). If the foreign DNA that is introduced comes from a different species, the host organism is called transgenic. Bacteria, plants, and animals have been genetically modified since the early 1970s for academic, medical, agricultural, and industrial purposes. In the US, GMOs such as Roundup-ready soybeans and borer-resistant corn are part of many common processed foods.
Gene Targeting
Although classical methods of studying the function of genes began with a given phenotype and determined the genetic basis of that phenotype, modern techniques allow researchers to start at the DNA sequence level and ask: “What does this gene or DNA element do?” This technique, called reverse genetics, has resulted in reversing the classic genetic methodology. This method would be similar to damaging a body part to determine its function. An insect that loses a wing cannot fly, which means that the function of the wing is flight. The classical genetic method would compare insects that cannot fly with insects that can fly, and observe that the non-flying insects have lost wings. Similarly, mutating or deleting genes provides researchers with clues about gene function. The methods used to disable gene function are collectively called gene targeting. Gene targeting is the use of recombinant DNA vectors to alter the expression of a particular gene, either by introducing mutations in a gene, or by eliminating the expression of a certain gene by deleting a part or all of the gene sequence from the genome of an organism.
Biotechnology in Medicine and Agriculture
It is easy to see how biotechnology can be used for medicinal purposes. Knowledge of the genetic makeup of our species, the genetic basis of heritable diseases, and the invention of technology to manipulate and fix mutant genes provides methods to treat the disease. Biotechnology in agriculture can enhance resistance to disease, pest, and environmental stress, and improve both crop yield and quality.
Genetic Diagnosis and Gene Therapy
The process of testing for suspected genetic defects before administering treatment is called genetic diagnosis by genetic testing. Depending on the inheritance patterns of a disease-causing gene, family members are advised to undergo genetic testing. For example, women diagnosed with breast cancer are usually advised to have a biopsy so that the medical team can determine the genetic basis of cancer development. Treatment plans are based on the findings of genetic tests that determine the type of cancer. If the cancer is caused by inherited gene mutations, other female relatives are also advised to undergo genetic testing and periodic screening for breast cancer. Genetic testing is also offered for fetuses (or embryos with in vitro fertilization) to determine the presence or absence of disease-causing genes in families with specific debilitating diseases.
Gene therapy is a genetic engineering technique used to cure disease. In its simplest form, it involves the introduction of a good gene at a random location in the genome to aid the cure of a disease that is caused by a mutated gene. The good gene is usually introduced into diseased cells as part of a vector transmitted by a virus that can infect the host cell and deliver the foreign DNA (Figure 14.49). More advanced forms of gene therapy try to correct the mutation at the original site in the genome, such as is the case with treatment of severe combined immunodeficiency (SCID).
Figure 9.49 Gene therapy using an adenovirus vector can be used to cure certain genetic diseases in which a person has a defective gene. (credit: NIH)
Production of Vaccines, Antibiotics, and Hormones
Traditional vaccination strategies use weakened or inactive forms of microorganisms to mount the initial immune response. Modern techniques use the genes of microorganisms cloned into vectors to mass produce the desired antigen. The antigen is then introduced into the body to stimulate the primary immune response and trigger immune memory. Genes cloned from the influenza virus have been used to combat the constantly changing strains of this virus.
Antibiotics are a biotechnological product. They are naturally produced by microorganisms, such as fungi, to attain an advantage over bacterial populations. Antibiotics are produced on a large scale by cultivating and manipulating fungal cells.
Recombinant DNA technology was used to produce large-scale quantities of human insulin in E. coli as early as 1978. Previously, it was only possible to treat diabetes with pig insulin, which caused allergic reactions in humans because of differences in the gene product. In addition, human growth hormone (HGH) is used to treat growth disorders in children. The HGH gene was cloned from a cDNA library and inserted into E. coli cells by cloning it into a bacterial vector.
Transgenic Animals
Although several recombinant proteins used in medicine are successfully produced in bacteria, some proteins require a eukaryotic animal host for proper processing. For this reason, the desired genes are cloned and expressed in animals, such as sheep, goats, chickens, and mice. Animals that have been modified to express recombinant DNA are called transgenic animals. Several human proteins are expressed in the milk of transgenic sheep and goats, and some are expressed in the eggs of chickens. Mice have been used extensively for expressing and studying the effects of recombinant genes and mutations.
Transgenic Plants
Manipulating the DNA of plants (i.e., creating GMOs) has helped to create desirable traits, such as disease resistance, herbicide and pesticide resistance, better nutritional value, and better shelf-life (Figure 14.50). Plants are the most important source of food for the human population. Farmers developed ways to select for plant varieties with desirable traits long before modern-day biotechnology practices were established.

Figure 9.50 Corn, a major agricultural crop used to create products for a variety of industries, is often modified through plant biotechnology. (credit: Keith Weller, USDA)
Plants that have received recombinant DNA from other species are called transgenic plants. Because they are not natural, transgenic plants and other GMOs are closely monitored by government agencies to ensure that they are fit for human consumption and do not endanger other plant and animal life. Because foreign genes can spread to other species in the environment, extensive testing is required to ensure ecological stability. Staples like corn, potatoes, and tomatoes were the first crop plants to be genetically engineered.
Transformation of Plants Using Agrobacterium tumefaciens
Gene transfer occurs naturally between species in microbial populations. Many viruses that cause human diseases, such as cancer, act by incorporating their DNA into the human genome. In plants, tumors caused by the bacterium Agrobacterium tumefaciens occur by transfer of DNA from the bacterium to the plant. Although the tumors do not kill the plants, they make the plants stunted and more susceptible to harsh environmental conditions. Many plants, such as walnuts, grapes, nut trees, and beets, are affected by A. tumefaciens. The artificial introduction of DNA into plant cells is more challenging than in animal cells because of the thick plant cell wall.
Researchers used the natural transfer of DNA from Agrobacterium to a plant host to introduce DNA fragments of their choice into plant hosts. In nature, the disease-causing A. tumefaciens have a set of plasmids, called the Ti plasmids (tumor-inducing plasmids), that contain genes for the production of tumors in plants. DNA from the Ti plasmid integrates into the infected plant cell’s genome. Researchers manipulate the Ti plasmids to remove the tumor-causing genes and insert the desired DNA fragment for transfer into the plant genome. The Ti plasmids carry antibiotic resistance genes to aid selection and can be propagated in E. coli cells as well.
The Organic Insecticide Bacillus thuringiensis
Bacillus thuringiensis (Bt) is a bacterium that produces protein crystals during sporulation that are toxic to many insect species that affect plants. Bt toxin has to be ingested by insects for the toxin to be activated. Insects that have eaten Bt toxin stop feeding on the plants within a few hours. After the toxin is activated in the intestines of the insects, death occurs within a couple of days. Modern biotechnology has allowed plants to encode their own crystal Bt toxin that acts against insects. The crystal toxin genes have been cloned from Bt and introduced into plants. Bt toxin has been found to be safe for the environment, non-toxic to humans and other mammals, and is approved for use by organic farmers as a natural insecticide.
Flavr Savr Tomato
The first GM crop to be introduced into the market was the Flavr Savr Tomato produced in 1994. Antisense RNA technology was used to slow down the process of softening and rotting caused by fungal infections, which led to increased shelf life of the GM tomatoes. Additional genetic modification improved the flavor of this tomato. The Flavr Savr tomato did not successfully stay in the market because of problems maintaining and shipping the crop.
9.19 | Mapping Genomes
Genomics is the study of entire genomes, including the complete set of genes, their nucleotide sequence and organization, and their interactions within a species and with other species. Genome mapping is the process of finding the locations of genes on each chromosome. The maps created by genome mapping are comparable to the maps that we use to navigate streets. A genetic map is an illustration that lists genes and their location on a chromosome. Genetic maps provide the big picture (similar to a map of interstate highways) and use genetic markers (similar to landmarks). A genetic marker is a gene or sequence on a chromosome that co-segregates (shows genetic linkage) with a specific trait. Early geneticists called this linkage analysis. Physical maps present the intimate details of smaller regions of the chromosomes (similar to a detailed road map). A physical map is a representation of the physical distance, in nucleotides, between genes or genetic markers. Both genetic linkage maps and physical maps are required to build a complete picture of the genome. Having a complete map of the genome makes it easier for researchers to study individual genes. Human genome maps help researchers in their efforts to identify human disease-causing genes related to illnesses like cancer, heart disease, and cystic fibrosis. Genome mapping can be used in a variety of other applications, such as using live microbes to clean up pollutants or even prevent pollution. Research involving plant genome mapping may lead to producing higher crop yields or developing plants that better adapt to climate change.
Genetic Maps
The study of genetic maps begins with linkage analysis, a procedure that analyzes the recombination frequency between genes to determine if they are linked or show independent assortment. The term linkage was used before the discovery of DNA. Early geneticists relied on the observation of phenotypic changes to understand the genotype of an organism. Shortly after Gregor Mendel (the father of modern genetics) proposed that traits were determined by what are now known as genes, other researchers observed that different traits were often inherited together, and thereby deduced that the genes were physically linked by being located on the same chromosome. The mapping of genes relative to each other based on linkage analysis led to the development of the first genetic maps.
Observations that certain traits were always linked and certain others were not linked came from studying the offspring of crosses between parents with different traits. For example, in experiments performed on the garden pea, it was discovered that the color of the flower and shape of the plant’s pollen were linked traits, and therefore the genes encoding these traits were in close proximity on the same chromosome. The exchange of DNA between homologous pairs of chromosomes is called genetic recombination, which occurs by the crossing over of DNA between homologous strands of DNA, such as nonsister chromatids. Linkage analysis involves studying the recombination frequency between any two genes. The greater the distance between two genes, the higher the chance that a recombination event will occur between them, and the higher the recombination frequency between them. Two possibilities for recombination between two nonsister chromatids during meiosis are shown in Figure 9.51. If the recombination frequency between two genes is less than 50 percent, they are said to be linked.
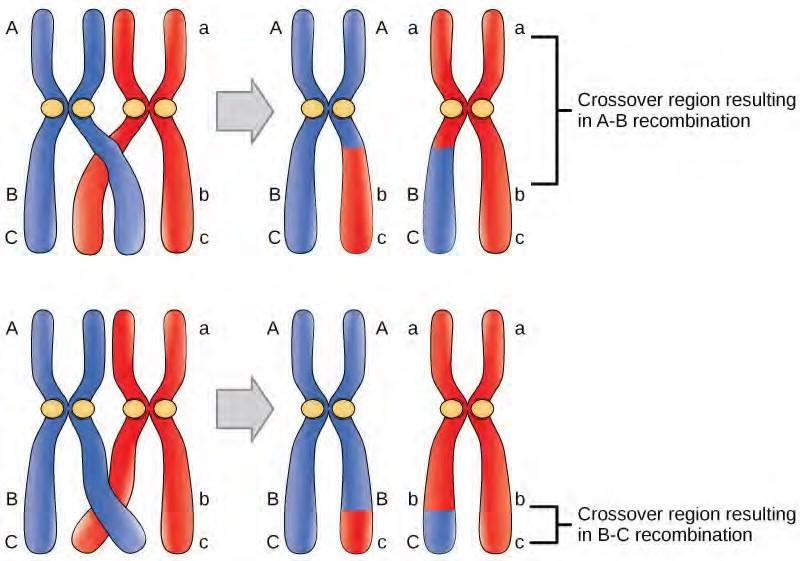
Figure 9.51 Crossover may occur at different locations on the chromosome. Recombination between genes A and B is more frequent than recombination between genes B and C because genes A and B are farther apart; a crossover is therefore more likely to occur between them.
The generation of genetic maps requires markers, just as a road map requires landmarks (such as rivers and mountains). Early genetic maps were based on the use of known genes as markers. More sophisticated markers, including those based on non-coding DNA, are now used to compare the genomes of individuals in a population. Although individuals of a given species are genetically similar, they are not identical; every individual has a unique set of traits. These minor differences in the genome between individuals in a population are useful for the purposes of genetic mapping. In general, a good genetic marker is a region on the chromosome that shows variability or polymorphism (multiple forms) in the population.
Some genetic markers used in generating genetic maps are restriction fragment length polymorphisms (RFLP), variable number of tandem repeats (VNTRs), microsatellite polymorphisms, and the single nucleotide polymorphisms (SNPs). RFLPs (sometimes pronounced “rif-lips”) are detected when the DNA of an individual is cut with a restriction endonuclease that recognizes specific sequences in the
DNA to generate a series of DNA fragments, which are then analyzed by gel electrophoresis. The DNA of every individual will give rise to a unique pattern of bands when cut with a particular set of restriction endonucleases; this is sometimes referred to as an individual’s DNA “fingerprint.” Certain regions of the chromosome that are subject to polymorphism will lead to the generation of the unique banding pattern. VNTRs are repeated sets of nucleotides present in the non-coding regions of DNA. Non-coding, or “junk,” DNA has no known biological function; however, research shows that much of this DNA is actually transcribed. While its function is uncertain, it is certainly active, and it may be involved in the regulation of coding genes. The number of repeats may vary in individual organisms of a population. Microsatellite polymorphisms are similar to VNTRs, but the repeat unit is very small. SNPs are variations in a single nucleotide.
Because genetic maps rely completely on the natural process of recombination, mapping is affected by natural increases or decreases in the level of recombination in any given area of the genome. Some parts of the genome are recombination hotspots, whereas others do not show a propensity for recombination. For this reason, it is important to look at mapping information developed by multiple methods.
Physical Maps
A physical map provides detail of the actual physical distance between genetic markers, as well as the number of nucleotides. There are three methods used to create a physical map: cytogenetic mapping, radiation hybrid mapping, and sequence mapping. Cytogenetic mapping uses information obtained by microscopic analysis of stained sections of the chromosome (Figure 9.52). It is possible to determine the approximate distance between genetic markers using cytogenetic mapping, but not the exact distance (number of base pairs). Radiation hybrid mapping uses radiation, such as x-rays, to break the DNA into fragments. The amount of radiation can be adjusted to create smaller or larger fragments. This technique overcomes the limitation of genetic mapping and is not affected by increased or decreased recombination frequency. Sequence mapping resulted from DNA sequencing technology that allowed for the creation of detailed physical maps with distances measured in terms of the number of base pairs. The creation of genomic libraries and complementary DNA (cDNA) libraries (collections of cloned sequences or all DNA from a genome) has sped up the process of physical mapping. A genetic site used to generate a physical map with sequencing technology (a sequence-tagged site, or STS) is a unique sequence in the genome with a known exact chromosomal location. An expressed sequence tag (EST) and a single sequence length polymorphism (SSLP) are common STSs. An EST is a short STS that is identified with cDNA libraries, while SSLPs are obtained from known genetic markers and provide a link between genetic maps and physical maps.
Figure 9.52 A cytogenetic map shows the appearance of a chromosome after it is stained and examined under a microscope. (credit: National Human Genome Research Institute)
Online Mendelian Inheritance in Man (OMIM) is a searchable online catalog of human genes and genetic disorders. This website shows genome mapping information, and also details the history and research of each trait and disorder. Click this link (http://openstaxcollege.org/l/OMIM) to search for traits (such as handedness) and genetic disorders (such as diabetes).
9.20 | Whole-Genome Sequencing
Although there have been significant advances in the medical sciences in recent years, doctors are still confounded by some diseases, and they are using whole-genome sequencing to get to the bottom of the problem. Whole-genome sequencing is a process that determines the DNA sequence of an entire genome. Whole-genome sequencing is a brute-force approach to problem solving when there is a genetic basis at the core of a disease. Several laboratories now provide services to sequence, analyze, and interpret entire genomes.
For example, whole-exome sequencing is a lower-cost alternative to whole genome sequencing. In exome sequencing, only the coding, exon-producing regions of the DNA are sequenced. In 2010, whole exome sequencing was used to save a young boy whose intestines had multiple mysterious abscesses. The child had several colon operations with no relief. Finally, whole-exome sequencing was performed, which revealed a defect in a pathway that controls apoptosis (programmed cell death). A bone-marrow transplant was used to overcome this genetic disorder, leading to a cure for the boy. He was the first person to be successfully treated based on a diagnosis made by whole-exome sequencing. Today, human genome sequencing is more readily available and can be completed in a day or two for about $1000.
A sequence alignment is an arrangement of proteins, DNA, or RNA; it is used to identify regions of similarity between cell types or species, which may indicate conservation of function or structures. Sequence alignments may be used to construct phylogenetic trees. The following website uses a software program called BLAST (basic local alignment search tool) (http://blast.ncbi.nlm.nih.gov/Blast.cgi) .
Use of Whole-Genome Sequences of Model Organisms
The first genome to be completely sequenced was of a bacterial virus, the bacteriophage fx174 (5368 base pairs); this was accomplished by Fred Sanger using shotgun sequencing. Several other organelle and viral genomes were later sequenced. The first organism whose genome was sequenced was the bacterium Haemophilus influenzae; this was accomplished by Craig Venter in the 1980s. Approximately 74 different laboratories collaborated on the sequencing of the genome of the yeast Saccharomyces cerevisiae, which began in 1989 and was completed in 1996, because it was 60 times bigger than any other genome that had been sequenced. By 1997, the genome sequences of two important model organisms were available: the bacterium Escherichia coli K12 and the yeast Saccharomyces cerevisiae. Genomes of other model organisms, such as the mouse Mus musculus, the fruit fly Drosophila melanogaster, the nematode Caenorhabditis. elegans, and humans Homo sapiens are now known. A lot of basic research is performed in model organisms because the information can be applied to genetically similar organisms. A model organism is a species that is studied as a model to understand the biological processes in other species represented by the model organism. Having entire genomes sequenced helps with the research efforts in these model organisms. The process of attaching biological information to gene sequences is called genome annotation. Annotation of gene sequences helps with basic experiments in molecular biology, such as designing PCR primers and RNA targets.
Click through each step of genome sequencing at this site (http://openstaxcollege.org/l/ DNA_sequence) .
Uses of Genome Sequences
DNA microarrays are methods used to detect gene expression by analyzing an array of DNA fragments that are fixed to a glass slide or a silicon chip to identify active genes and identify sequences. Almost one million genotypic abnormalities can be discovered using microarrays, whereas whole-genome sequencing can provide information about all six billion base pairs in the human genome. Although the study of medical applications of genome sequencing is interesting, this discipline tends to dwell on abnormal gene function. Knowledge of the entire genome will allow future onset diseases and other genetic disorders to be discovered early, which will allow for more informed decisions to be made about lifestyle, medication, and having children. Genomics is still in its infancy, although someday it may become routine to use whole-genome sequencing to screen every newborn to detect genetic abnormalities.
In addition to disease and medicine, genomics can contribute to the development of novel enzymes that convert biomass to biofuel, which results in higher crop and fuel production, and lower cost to the consumer. This knowledge should allow better methods of control over the microbes that are used in the production of biofuels. Genomics could also improve the methods used to monitor the impact of pollutants on ecosystems and help clean up environmental contaminants. Genomics has allowed for the development of agrochemicals and pharmaceuticals that could benefit medical science and agriculture.
It sounds great to have all the knowledge we can get from whole-genome sequencing; however, humans have a responsibility to use this knowledge wisely. Otherwise, it could be easy to misuse the power of such knowledge, leading to discrimination based on a person’s genetics, human genetic engineering, and other ethical concerns. This information could also lead to legal issues regarding health and privacy.
9.21 | Applying Genomics
The introduction of DNA sequencing and whole genome sequencing projects, particularly the Human Genome project, has expanded the applicability of DNA sequence information. Genomics is now being used in a wide variety of fields, such as metagenomics, pharmacogenomics, and mitochondrial genomics. The most commonly known application of genomics is to understand and find cures for diseases.
Predicting Disease Risk at the Individual Level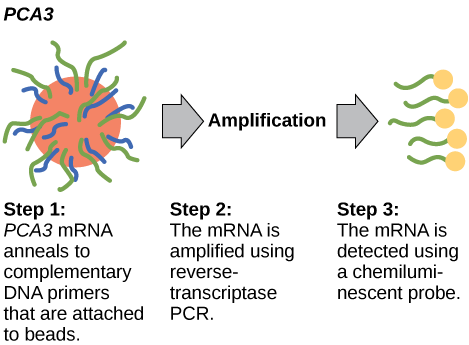
Predicting the risk of disease involves screening currently healthy individuals by genome analysis at the individual level. Intervention with lifestyle changes and drugs can be recommended before disease onset. However, this approach is most applicable when the problem resides within a single gene defect. Such defects only account for approximately 5 percent of diseases in developed countries. Most of the common diseases, such as heart disease, are multi-factored or polygenic, which is a phenotypic characteristic that involves two or more genes, and also involve environmental factors such as diet. In April 2010, scientists at Stanford University published the genome analysis of a healthy individual (Stephen Quake, a scientist at Stanford University, who had his genome sequenced); the analysis predicted his propensity to acquire various diseases. A risk assessment was performed to analyze Quake’s percentage of risk for 55 different medical conditions. A rare genetic mutation was found, which showed him to be at risk for sudden heart attack. He was also predicted to have a 23 percent risk of developing prostate cancer and a 1.4 percent risk of developing Alzheimer’s. The scientists used databases and several publications to analyze the genomic data. Even though genomic sequencing is becoming more affordable and analytical tools are becoming more reliable, ethical issues surrounding genomic analysis at a population level remain to be addressed.
Figure 14.55 PCA3 is a gene that is expressed in prostate epithelial cells and overexpressed in cancerous cells. A high concentration of PCA3 in urine is indicative of prostate cancer. The PCA3 test is considered to be a better indicator of cancer than the more well know PSA test, which measures the level of PSA (prostate-specific antigen) in the blood.
In 2011, the United States Preventative Services Task Force recommended against using the PSA test to screen healthy men for prostate cancer. Their recommendation is based on evidence that screening does not reduce the risk of death from prostate cancer. Prostate cancer often develops very slowly and does not cause problems, while the cancer treatment can have severe side effects. The PCA3 test is considered to be more accurate, but screening may still result in men who would not have been harmed by the cancer itself suffering side effects from treatment. What do you think? Should all healthy men be screened for prostate cancer using the PCA3 or PSA test? Should people in general be screened to find out if they have a genetic risk for cancer or other diseases?
Pharmacogenomics and Toxicogenomics
Pharmacogenomics, also called toxicogenomics, involves evaluating the effectiveness and safety of drugs on the basis of information from an individual’s genomic sequence. Genomic responses to drugs can be studied using experimental animals (such as laboratory rats or mice) or live cells in the laboratory before embarking on studies with humans. Studying changes in gene expression could provide information about the transcription profile in the presence of the drug, which can be used as an early indicator of the potential for toxic effects. For example, genes involved in cellular growth and controlled cell death, when disturbed, could lead to the growth of cancerous cells. Genome-wide studies can also help to find new genes involved in drug toxicity. Personal genome sequence information can be used to prescribe medications that will be most effective and least toxic on the basis of the individual patient’s genotype. The gene signatures may not be completely accurate, but can be tested further before pathologic symptoms arise.
Microbial Genomics: Metagenomics
Traditionally, microbiology has been taught with the view that microorganisms are best studied under pure culture conditions, which involves isolating a single type of cell and culturing it in the laboratory. Because microorganisms can go through several generations in a matter of hours, their gene expression profiles adapt to the new laboratory environment very quickly. In addition, the vast majority of bacterial species resist being cultured in isolation. Most microorganisms do not live as isolated entities, but in microbial communities known as biofilms. For all of these reasons, pure culture is not always the best way to study microorganisms. Metagenomics is the study of the collective genomes of multiple species that grow and interact in an environmental niche. Metagenomics can be used to identify new species more rapidly and to analyze the effect of pollutants on the environment (Figure 14.56).
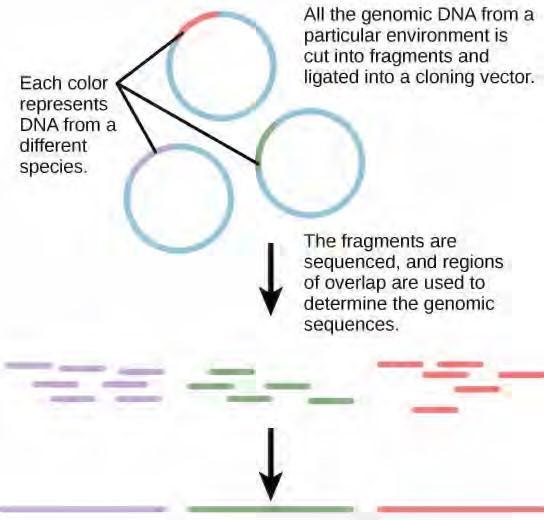
Figure 14.56 Metagenomics involves isolating DNA from multiple species within an environmental niche.
Microbial Genomics: Creation of New Biofuels
Knowledge of the genomics of microorganisms is being used to find better ways to harness biofuels from algae and cyanobacteria. The primary sources of fuel today are coal, oil, wood, and other plant products, such as ethanol. Although plants are renewable resources, there is still a need to find more alternative renewable sources of energy to meet our population’s energy demands. The microbial world is one of the largest resources for genes that encode new enzymes and produce new organic compounds, and it remains largely untapped. Microorganisms are used to create products, such as enzymes that are used in research, antibiotics, and other anti-microbial mechanisms. Microbial genomics is helping to develop diagnostic tools, improved vaccines, new disease treatments, and advanced environmental cleanup techniques.
Mitochondrial Genomics
Mitochondria are intracellular organelles that contain their own DNA. Mitochondrial DNA mutates at a rapid rate and is often used to study evolutionary relationships. Another feature that makes studying the mitochondrial genome interesting is that the mitochondrial DNA in most multicellular organisms is passed on from the mother during the process of fertilization. For this reason, mitochondrial genomics is often used to trace genealogy.
Information and clues obtained from DNA samples found at crime scenes have been used as evidence in court cases, and genetic markers have been used in forensic analysis. Genomic analysis has also become useful in this field. In 2001, the first use of genomics in forensics was published. It was a collaborative attempt between academic research institutions and the FBI to solve the mysterious cases of anthrax communicated via the US Postal Service. Using microbial genomics, researchers determined that a specific strain of anthrax was used in all the mailings.
Genomics in Agriculture
Genomics can reduce the trials and failures involved in scientific research to a certain extent, which could improve the quality and quantity of crop yields in agriculture. Linking traits to genes or gene signatures helps to improve crop breeding to generate hybrids with the most desirable qualities. Scientists use genomic data to identify desirable traits, and then transfer those traits to a different organism. Scientists are discovering how genomics can improve the quality and quantity of agricultural production. For example, scientists could use desirable traits to create a useful product or enhance an existing product, such as making a drought-sensitive crop more tolerant of the dry season.
9.22 | Genomics and Proteomics
Proteins are the final products of genes, which help perform the function encoded by the gene. Proteins are composed of amino acids and play important roles in the cell. All enzymes (except ribozymes) are proteins that act as catalysts to affect the rate of reactions. Proteins are also regulatory molecules, and some are hormones. Transport proteins, such as hemoglobin, help transport oxygen to various organs. Antibodies that defend against foreign particles are also proteins. In the diseased state, protein function can be impaired because of changes at the genetic level or because of direct impact on a specific protein.
A proteome is the entire set of proteins produced by a cell type. Proteomes can be studied using the knowledge of genomes because genes code for mRNAs, and the mRNAs encode proteins. Although mRNA analysis is a step in the right direction, not all mRNAs are translated into proteins. The study of the function of proteomes is called proteomics. Proteomics complements genomics and is useful when scientists want to test their hypotheses that were based on genes. Even though all cells of a multicellular organism have the same set of genes, the set of proteins produced in different tissues is different and dependent on gene expression. Thus, the genome is constant, but the proteome varies and is dynamic within an organism. In addition, RNAs can be alternately spliced (cut and pasted to create novel combinations and novel proteins) and many proteins are modified after translation by processes such as proteolytic cleavage, phosphorylation, glycosylation, and ubiquitination. There are also protein-protein interactions, which complicate the study of proteomes. Although the genome provides a blueprint, the final architecture depends on several factors that can change the progression of events that generate the proteome.
Metabolomics is related to genomics and proteomics. Metabolomics involves the study of small molecule metabolites found in an organism. The metabolome is the complete set of metabolites that are related to the genetic makeup of an organism. Metabolomics offers an opportunity to compare genetic makeup and physical characteristics, as well as genetic makeup and environmental factors. The goal of metabolome research is to identify, quantify, and catalogue all of the metabolites that are found in the tissues and fluids of living organisms.
Cancer Proteomics
Genomes and proteomes of patients suffering from specific diseases are being studied to understand the genetic basis of the disease. The most prominent disease being studied with proteomic approaches is cancer. Proteomic approaches are being used to improve screening and early detection of cancer; this is achieved by identifying proteins whose expression is affected by the disease process. An individual protein is called a biomarker, whereas a set of proteins with altered expression levels is called a protein signature. For a biomarker or protein signature to be useful as a candidate for early screening and detection of a cancer, it must be secreted in body fluids, such as sweat, blood, or urine, such that large scale screenings can be performed in a non-invasive fashion. The current problem with using biomarkers for the early detection of cancer is the high rate of false-negative results. A false negative is an incorrect test result that should have been positive. In other words, many cases of cancer go undetected, which makes biomarkers unreliable. Some examples of protein biomarkers used in cancer detection are CA-125 for ovarian cancer and PSA for prostate cancer. Protein signatures may be more reliable than biomarkers to detect cancer cells. Proteomics is also being used to develop individualized treatment plans, which involves the prediction of whether or not an individual will respond to specific drugs and the side effects that the individual may experience. Proteomics is also being used to predict the possibility of disease recurrence.
The National Cancer Institute has developed programs to improve the detection and treatment of cancer. The Clinical Proteomic Technologies for Cancer and the Early Detection Research Network are efforts to identify protein signatures specific to different types of cancers. The Biomedical Proteomics Program is designed to identify protein signatures and design effective therapies for cancer patients.
REVIEW QUESTIONS
The AUC and AUA codons in mRNA both specify isoleucine. What feature of the genetic code explains this?
complementarity
nonsense codons
universality
degeneracy
How many nucleotides are in 12 mRNA codons?
12
24
36
48
The -10 and -35 regions of prokaryotic promoters are called consensus sequences because ________.
they are identical in all bacterial
species
they are similar in all bacterial species
they exist in all organisms
they have the same function in all organisms
Which feature of promoters can be found in both prokaryotes and eukaryotes?
GC box
TATA box
octamer box
-10 and -35 sequences
The RNA components of ribosomes are synthesized in the ________.
cytoplasm
nucleus
nucleolus
endoplasmic reticulum
In any given species, there are at least how many types of aminoacyl tRNA synthetases?
20
40
100
Control of gene expression in eukaryotic cells occurs at which level(s)?
only the transcriptional level
epigenetic and transcriptional levels
epigenetic, transcriptional, and translational
levels
epigenetic, transcriptional, post-transcriptional,
translational, and post-translational levels
Post-translational control refers to:
regulation of gene expression after transcription
regulation of gene expression after translation
control of epigenetic activation
period between transcription and translation
If glucose is absent, but so is lactose, the lac operon will be ________.
activated
repressed
activated, but only partially
mutated
Prokaryotic cells lack a nucleus. Therefore, the genes in prokaryotic cells are:
all expressed, all of the time
transcribed and translated almost simultaneously
transcriptionally controlled because translation
begins before transcription ends
What will result from the binding of a transcription factor to an enhancer region?
decreased transcription of an adjacent gene
increased transcription of a distant gene
alteration of the translation of an adjacent gene
initiation of the recruitment of RNA polymerase
Which of the following are involved in post- transcriptional control?
control of RNA splicing
control of RNA shuttling
control of RNA stability
Binding of an RNA binding protein will ________ the stability of the RNA molecule.
increase
decrease
neither increase nor decrease
either increase or decrease
Post-translational modifications of proteins can affect which of the following?
protein function
transcriptional regulation
chromatin modification
all of the above
Cancer causing genes are called ________.
transformation genes
tumor suppressor genes
oncogenes
mutated genes
Targeted therapies are used in patients with a set gene expression pattern. A targeted therapy that prevents the activation of the estrogen receptor in breast cancer would be beneficial to which type of patient?
patients who express the EGFR receptor in normal cells
patients with a mutation that inactivates the estrogen receptor
patients with lots of the estrogen receptor expressed in their tumor
patients that have no estrogen receptor expressed in their tumor
GMOs are created by ________.
generating genomic DNA fragments with restriction endonucleases
introducing recombinant DNA into an organism by any means
overexpressing proteins in E. coli.
all of the above
Gene therapy can be used to introduce foreign DNA into cells ________.
for molecular cloning
by PCR
of tissues to cure inheritable disease
all of the above
Insulin produced by molecular cloning:
is of pig origin
is a recombinant protein
is made by the human pancreas
is recombinant DNA
CRITICAL THINKING QUESTIONS
Imagine if there were 200 commonly occurring amino acids instead of 20. Given what you know about the genetic code, what would be the shortest possible codon length? Explain.
Discuss how degeneracy of the genetic code makes cells more robust to mutations.
If mRNA is complementary to the DNA template strand and the DNA template strand is complementary to the DNA nontemplate strand, then why are base sequences of mRNA and the DNA nontemplate strand not identical? Could they ever be?
In your own words, describe the difference between rho-dependent and rho-independent termination of transcription in prokaryotes.
Transcribe and translate the following DNA sequence (nontemplate strand): 5′-ATGGCCGGTTATTAAGCA-3′
Explain how single nucleotide changes can have vastly different effects on protein
Name two differences between prokaryotic and eukaryotic cells and how these differences benefit multicellular organisms.
Describe how controlling gene expression will alter the overall protein levels in the cell.
Hypothetically, how could you reverse this process to turn these genes back on?
A mutation within the promoter region can alter transcription of a gene. Describe how this can happen.
What could happen if a cell had too much of an activating transcription factor present?
Describe how RBPs can prevent miRNAs from degrading an RNA molecule.
How can external stimuli alter post-transcriptional control of gene expression?
Protein modification can alter gene expression in many ways. Describe how phosphorylation of proteins can alter gene expression.
Alternative forms of a protein can be beneficial or harmful to a cell. What do you think would happen if too much of an alternative protein bound to the 3′ UTR of an RNA and caused it to degrade?
Changes in epigenetic modifications alter the accessibility and transcription of DNA. Describe how environmental stimuli, such as ultraviolet light exposure, could modify gene expression.
New drugs are being developed that decrease DNA methylation and prevent the removal of acetyl groups from histone proteins. Explain how these drugs could affect gene expression to help kill tumor cells.
How can understanding the gene expression pattern in a cancer cell tell you something about that specific form of cancer?
KEY TERMS
antibiotic resistance ability of an organism to be unaffected by the actions of an antibiotic
biomarker individual protein that is uniquely produced in a diseased state
biotechnology use of biological agents for technological advancement
clone exact replica
deoxynucleotide individual monomer (single unit) of DNA
gel electrophoresis technique used to separate molecules on the basis of size using electric charge
gene targeting method for altering the sequence of a specific gene by introducing the modified version on a vector
gene therapy technique used to cure inheritable diseases by replacing mutant genes with good genes
genetic diagnosis diagnosis of the potential for disease development by analyzing disease-causing genes
genetic engineering alteration of the genetic makeup of an organism
genetic map outline of genes and their location on a chromosome
genetic marker gene or sequence on a chromosome with a known location that is associated with a
specific trait
genetic recombination exchange of DNA between homologous pairs of chromosomes
genetic testing process of testing for the presence of disease-causing genes
genetically modified organism (GMO) organism whose genome has been artificially changed
genomics study of entire genomes including the complete set of genes, their nucleotide sequence and organization, and their interactions within a species and with other species
host DNA DNA that is present in the genome of the organism of interest
linkage analysis procedure that analyzes the recombination of genes to determine if they are linked
proteomics study of the function of proteomes
recombinant DNA combination of DNA fragments generated by molecular cloning that does not exist in nature; also known as a chimeric molecule
recombinant protein protein product of a gene derived by molecular cloning
reproductive cloning cloning of entire organisms
restriction endonuclease enzyme that can recognize and cleave specific DNA sequences
systems biology study of whole biological systems (genomes and proteomes) based on interactions within the system
transgenic organism that receives DNA from a different species
7-methylguanosine cap modification added to the 5′ end of pre-mRNAs to protect mRNA from degradation and assist translation
aminoacyl tRNA synthetase enzyme that “charges” tRNA molecules by catalyzing a bond between the tRNA and a corresponding amino acid anticodon three-nucleotide sequence in a tRNA molecule that corresponds to an mRNA codon
Central Dogma states that genes specify the sequence of mRNAs, which in turn specify the sequence of proteins
codon three consecutive nucleotides in mRNA that specify the insertion of an amino acid or the release of a polypeptide chain during translation
colinear in terms of RNA and protein, three “units” of RNA (nucleotides) specify one “unit” of protein (amino acid) in a consecutive fashion consensus DNA sequence that is used by many species to perform the same or similar functions
core enzyme prokaryotic RNA polymerase consisting of α, α, β, and β‘ but missing σ; this complex performs elongation
degeneracy (of the genetic code) describes that a given amino acid can be encoded by more than one nucleotide triplet; the code is degenerate, but not ambiguous
downstream nucleotides following the initiation site in the direction of mRNA transcription; in general, sequences that are toward the 3′ end relative to a site on the mRNA exon sequence present in protein-coding mRNA after completion of pre-mRNA splicing
FACT complex that “facilitates chromatin transcription” by disassembling nucleosomes ahead of a transcribing RNA polymerase II and reassembling them after the polymerase passes by
GC-rich box (GGCG) nonessential eukaryotic promoter sequence that binds cellular factors to increase the efficiency of transcription; may be present several times in a promoter
hairpin structure of RNA when it folds back on itself and forms intramolecular hydrogen bonds between complementary nucleotides
holoenzyme prokaryotic RNA polymerase consisting of α, α, β, β‘, and σ; this complex is
responsible for transcription initiation
initiation site nucleotide from which mRNA synthesis proceeds in the 5′ to 3′ direction; denoted with a “+1” initiator tRNA in prokaryotes, called tRNAMetf ; in eukaryotes, called tRNAi; a tRNA that interacts
with a start codon, binds directly to the ribosome P site, and links to a special methionine to begin a polypeptide chain intron non–protein-coding intervening sequences that are spliced from mRNA during processing
Kozak’s rules determines the correct initiation AUG in a eukaryotic mRNA; the following consensus sequence must appear around the AUG: 5’-GCC(purine)CCAUGG-3’; the bolded bases are most important nonsense codon one of the three mRNA codons that specifies termination of translation
nontemplate strand strand of DNA that is not used to transcribe mRNA; this strand is identical to the mRNA except that T nucleotides in the DNA are replaced by U nucleotides in the mRNA Octamer box (ATTTGCAT) nonessential eukaryotic promoter sequence that binds cellular factors to increase the efficiency of transcription; may be present several times in a promoter
peptidyl transferase RNA-based enzyme that is integrated into the 50S ribosomal subunit and catalyzes the formation of peptide bonds
plasmid extrachromosomal, covalently closed, circular DNA molecule that may only contain one or a few genes; common in prokaryotes
poly-A tail modification added to the 3′ end of pre-mRNAs to protect mRNA from degradation and assist mRNA export from the nucleus
polysome mRNA molecule simultaneously being translated by many ribosomes all going in the same direction
preinitiation complex cluster of transcription factors and other proteins that recruit RNA polymerase II for transcription of a DNA template
promoter DNA sequence to which RNA polymerase and associated factors bind and initiate transcription
reading frame sequence of triplet codons in mRNA that specify a particular protein; a ribosome shift of one or two nucleotides in either direction completely abolishes synthesis of that protein
Rho-dependent termination in prokaryotes, termination of transcription by an interaction between RNA polymerase and the rho protein at a run of G nucleotides on the DNA template
Rho-independent termination sequence-dependent termination of prokaryotic mRNA synthesis; caused by hairpin formation in the mRNA that stalls the polymerase
RNA editing direct alteration of one or more nucleotides in an mRNA that has already been synthesized
Shine-Dalgarno sequence (AGGAGG); initiates prokaryotic translation by interacting with rRNA molecules comprising the 30S ribosome signal sequence short tail of amino acids that directs a protein to a specific cellular compartment
small nuclear RNA molecules synthesized by RNA polymerase III that have a variety of functions, including splicing pre-mRNAs and regulating transcription factors splicing process of removing introns and reconnecting exons in a pre-mRNA
start codon AUG (or rarely, GUG) on an mRNA from which translation begins; always specifies methionine
TATA box conserved promoter sequence in eukaryotes and prokaryotes that helps to establish the
initiation site for transcription
template strand strand of DNA that specifies the complementary mRNA molecule transcription bubble region of locally unwound DNA that allows for transcription of mRNA
upstream nucleotides preceding the initiation site; in general, sequences toward the 5′ end relative to a site on the mRNA
3′ UTR 3′ untranslated region; region just downstream of the protein-coding region in an RNA molecule that is not translated
5′ cap a methylated guanosine triphosphate (GTP) molecule that is attached to the 5′ end of a messenger RNA to protect the end from degradation
5′ UTR 5′ untranslated region; region just upstream of the protein-coding region in an RNA molecule that is not translated
activator protein that binds to prokaryotic operators to increase transcription
catabolite activator protein (CAP) protein that complexes with cAMP to bind to the promoter sequences of operons that control sugar processing when glucose is not available
DNA methylation epigenetic modification that leads to gene silencing; commonly found in cancer cells
enhancer segment of DNA that is upstream, downstream, perhaps thousands of nucleotides away, or on another chromosome that influence the transcription of a specific gene
epigenetic heritable changes that do not involve changes in the DNA sequence
eukaryotic initiation factor-2 (eIF-2) protein that binds first to an mRNA to initiate translation
gene expression processes that control the turning on or turning off of a gene
initiation complex protein complex containing eIF2-2 that starts translation
lac operon operon in prokaryotic cells that encodes genes required for processing and intake of lactose
negative regulator protein that prevents transcription
operator region of DNA outside of the promoter region that binds activators or repressors that control gene expression in prokaryotic cells
operon collection of genes involved in a pathway that are transcribed together as a single mRNA in prokaryotic cells poly-A tail a series of adenine nucleotides that are attached to the 3′ end of an mRNA to protect the end from degradation positive regulator protein that increases transcription
repressor protein that binds to the operator of prokaryotic genes to prevent transcription RISC protein complex that binds along with the miRNA to the RNA to degrade it
RNA stability how long an RNA molecule will remain intact in the cytoplasm
transcription factor protein that binds to the DNA at the promoter or enhancer region and that influences transcription of a gene
transcriptional start site site at which transcription begins
trp operon series of genes necessary to synthesize tryptophan in prokaryotic cells
Chapter Summary
9.1 DNA Structure and Sequencing
The currently accepted model of the double-helix structure of DNA was proposed by Watson and Crick. Some of the salient features are that the two strands that make up the double helix are complementary and anti-parallel in nature. Deoxyribose sugars and phosphates form the backbone of the structure, and the nitrogenous bases are stacked inside. The diameter of the double helix, 2 nm, is uniform throughout. A purine always pairs with a pyrimidine; A pairs with T, and G pairs with C. One turn of the helix has ten base pairs. During cell division, each daughter cell receives a copy of the DNA by a process known as DNA replication. Prokaryotes are much simpler than eukaryotes in many of their features. Most prokaryotes contain a single, circular chromosome. In general, eukaryotic chromosomes contain a linear DNA molecule packaged into nucleosomes, and have two distinct regions that can be distinguished by staining, reflecting different states of packaging and compaction.
9.2 Basics of DNA Replication
The model for DNA replication suggests that the two strands of the double helix separate during replication, and each strand serves as a template from which the new complementary strand is copied. In conservative replication, the parental DNA is conserved, and the daughter DNA is newly synthesized. The semi-conservative method suggests that each of the two parental DNA strands acts as template for new DNA to be synthesized; after replication, each double-stranded DNA includes one parental or “old” strand and one “new” strand. The dispersive mode suggested that the two copies of the DNA would have segments of parental DNA and newly synthesized DNA.
9.3 DNA Replication in Prokaryotes
Replication in prokaryotes starts from a sequence found on the chromosome called the origin of replication—the point at which the DNA opens up. Helicase opens up the DNA double helix, resulting in the formation of the replication fork. Single-strand binding proteins bind to the single-stranded DNA near the replication fork to keep the fork open. Primase synthesizes an RNA primer to initiate synthesis by DNA polymerase, which can add nucleotides only in the 5′ to 3′ direction. One strand is synthesized continuously in the direction of the replication fork; this is called the leading strand. The other strand is synthesized in a direction away from the replication fork, in short stretches of DNA known as Okazaki fragments. This strand is known as the lagging strand. Once replication is completed, the RNA primers are replaced by DNA nucleotides and the DNA is sealed with DNA ligase, which creates phosphodiester bonds between the 3′-OH of one end and the 5′ phosphate of the other strand.
9.4 DNA Replication in Eukaryotes
Replication in eukaryotes starts at multiple origins of replication. The mechanism is quite similar to prokaryotes. A primer is required to initiate synthesis, which is then extended by DNA polymerase as it adds nucleotides one by one to the growing chain. The leading strand is synthesized continuously, whereas the lagging strand is synthesized in short stretches called Okazaki fragments. The RNA primers are replaced with DNA nucleotides; the DNA remains one continuous strand by linking the DNA fragments with DNA ligase. The ends of the chromosomes pose a problem as polymerase is unable to extend them without a primer. Telomerase, an enzyme with an inbuilt RNA template, extends the ends by copying the RNA template and extending one end of the chromosome. DNA polymerase can then extend the DNA using the primer. In this way, the ends of the chromosomes are protected.
9.5 DNA Repair
DNA polymerase can make mistakes while adding nucleotides. It edits the DNA by proofreading every newly added base. Incorrect bases are removed and replaced by the correct base, and then a new base is added. Most mistakes are corrected during replication, although when this does not happen, the mismatch repair mechanism is employed. Mismatch repair enzymes recognize the wrongly incorporated base and excise it from the DNA, replacing it with the correct base. In yet another type of repair, nucleotide excision repair, the incorrect base is removed along with a few bases on the 5′ and 3′ end, and these are replaced by copying the template with the help of DNA polymerase. The ends of the newly synthesized fragment are attached to the rest of the DNA using DNA ligase, which creates a phosphodiester bond.
Most mistakes are corrected, and if they are not, they may result in a mutation defined as a permanent change in the DNA sequence. Mutations can be of many types, such as substitution, deletion, insertion, and translocation. Mutations in repair genes may lead to serious consequences such as cancer. Mutations can be induced or may occur spontaneously.
9.6 The Genetic Code
The genetic code refers to the DNA alphabet (A, T, C, G), the RNA alphabet (A, U, C, G), and the polypeptide alphabet (20 amino acids). The Central Dogma describes the flow of genetic information in the cell from genes to mRNA to proteins. Genes are used to make mRNA by the process of transcription; mRNA is used to synthesize proteins by the process of translation. The genetic code is degenerate because 64 triplet codons in mRNA specify only 20 amino acids and three nonsense codons. Almost every species on the planet uses the same genetic code.
9.7 Prokaryotic Transcription
In prokaryotes, mRNA synthesis is initiated at a promoter sequence on the DNA template comprising two consensus sequences that recruit RNA polymerase. The prokaryotic polymerase consists of a core enzyme of four protein subunits and a σ protein that assists only with initiation. Elongation synthesizes mRNA in the 5′ to 3′ direction at a rate of 40 nucleotides per second. Termination liberates the mRNA and occurs either by rho protein interaction or by the formation of an mRNA hairpin.
9.8 Eukaryotic Transcription
Transcription in eukaryotes involves one of three types of polymerases, depending on the gene being transcribed. RNA polymerase II transcribes all of the protein-coding genes, whereas RNA polymerase I transcribes rRNA genes, and RNA polymerase III transcribes rRNA, tRNA, and small nuclear RNA genes. The initiation of transcription in eukaryotes involves the binding of several transcription factors to complex promoter sequences that are usually located upstream of the gene being copied. The mRNA is synthesized in the 5′ to 3′ direction, and the FACT complex moves and reassembles nucleosomes as the polymerase passes by. Whereas RNA polymerases I and III terminate transcription by protein- or RNA hairpin-dependent methods, RNA polymerase II transcribes for 1,000 or more nucleotides beyond the gene template and cleaves the excess during pre-mRNA processing.
9.9 RNA Processing in Eukaryotes
Eukaryotic pre-mRNAs are modified with a 5′ methylguanosine cap and a poly-A tail. These structures protect the mature mRNA from degradation and help export it from the nucleus. Pre-mRNAs also undergo splicing, in which introns are removed and exons are reconnected with single-nucleotide accuracy. Only finished mRNAs that have undergone 5′ capping, 3′ polyadenylation, and intron splicing are exported from the nucleus to the cytoplasm. Pre-rRNAs and pre-tRNAs may be processed by intramolecular cleavage, splicing, methylation, and chemical conversion of nucleotides. Rarely, RNA editing is also performed to insert missing bases after an mRNA has been synthesized.
9.10 Ribosomes and Protein Synthesis
The players in translation include the mRNA template, ribosomes, tRNAs, and various enzymatic factors. The small ribosomal subunit forms on the mRNA template either at the Shine-Dalgarno sequence (prokaryotes) or the 5′ cap (eukaryotes). Translation begins at the initiating AUG on the mRNA, specifying methionine. The formation of peptide bonds occurs between sequential amino acids specified by the mRNA template according to the genetic code. Charged tRNAs enter the ribosomal A site, and their amino acid bonds with the amino acid at the P site. The entire mRNA is translated in three-nucleotide “steps” of the ribosome. When a nonsense codon is encountered, a release factor binds and dissociates the components and frees the new protein. Folding of the protein occurs during and after translation.
9.11 Regulation of Gene Expression
While all somatic cells within an organism contain the same DNA, not all cells within that organism express the same proteins. Prokaryotic organisms express the entire DNA they encode in every cell, but not necessarily all at the same time. Proteins are expressed only when they are needed. Eukaryotic organisms express a subset of the DNA that is encoded in any given cell. In each cell type, the type and amount of protein is regulated by controlling gene expression. To express a protein, the DNA is first transcribed into RNA, which is then translated into proteins. In prokaryotic cells, these processes occur almost simultaneously. In eukaryotic cells, transcription occurs in the nucleus and is separate from the translation that occurs in the cytoplasm. Gene expression in prokaryotes is mostly regulated at the transcriptional level (some epigenetic and post-translational regulation is also present), whereas in eukaryotic cells, gene expression is regulated at the epigenetic, transcriptional, post-transcriptional, translational, and post-translational levels.
9.12 Prokaryotic Gene Regulation
The regulation of gene expression in prokaryotic cells occurs at the transcriptional level. There are three ways to control the transcription of an operon: repressive control, activator control, and inducible control. Repressive control, typified by the trp operon, uses proteins bound to the operator sequence to physically prevent the binding of RNA polymerase and the activation of transcription. Therefore, if tryptophan is not needed, the repressor is bound to the operator and transcription remains off. Activator control, typified by the action of CAP, increases the binding ability of RNA polymerase to the promoter when CAP is bound. In this case, low levels of glucose result in the binding of cAMP to CAP. CAP then binds the promoter, which allows RNA polymerase to bind to the promoter better. In the last example—the lac operon—two conditions must be met to initiate transcription. Glucose must not be present, and lactose must be available for the lac operon to be transcribed. If glucose is absent, CAP binds to the operator. If lactose is present, the repressor protein does not bind to its operator. Only when both conditions are met will RNA polymerase bind to the promoter to induce transcription.
9.13 Eukaryotic Epigenetic Gene Regulation
In eukaryotic cells, the first stage of gene expression control occurs at the epigenetic level. Epigenetic mechanisms control access to the chromosomal region to allow genes to be turned on or off. These mechanisms control how DNA is packed into the nucleus by regulating how tightly the DNA is wound around histone proteins. The addition or removal of chemical modifications (or flags) to histone proteins or DNA signals to the cell to open or close a chromosomal region. Therefore, eukaryotic cells can control whether a gene is expressed by controlling accessibility to transcription factors and the binding of RNA polymerase to initiate transcription.
9.14 Eukaryotic Transcription Gene Regulation
To start transcription, general transcription factors, such as TFIID, TFIIH, and others, must first bind to the TATA box and recruit RNA polymerase to that location. The binding of additional regulatory transcription factors to cis-acting elements will either increase or prevent transcription. In addition to promoter sequences, enhancer regions help augment transcription. Enhancers can be upstream, downstream, within a gene itself, or on other chromosomes. Transcription factors bind to enhancer regions to increase or prevent transcription.
9.15 Eukaryotic Post-transcriptional Gene Regulation
Post-transcriptional control can occur at any stage after transcription, including RNA splicing, nuclear shuttling, and RNA stability. Once RNA is transcribed, it must be processed to create a mature RNA that is ready to be translated. This involves the removal of introns that do not code for protein. Spliceosomes bind to the signals that mark the exon/intron border to remove the introns and ligate the exons together. Once this occurs, the RNA is mature and can be translated. RNA is created and spliced in the nucleus, but needs to be transported to the cytoplasm to be translated. RNA is transported to the cytoplasm through the nuclear pore complex. Once the RNA is in the cytoplasm, the length of time it resides there before being degraded, called RNA stability, can also be altered to control the overall amount of protein that is synthesized. The RNA stability can be increased, leading to longer residency time in the cytoplasm, or decreased, leading to shortened time and less protein synthesis. RNA stability is controlled by RNA-binding proteins (RPBs) and microRNAs (miRNAs). These RPBs and miRNAs bind to the 5′ UTR or the 3′ UTR of the RNA to increase or decrease RNA stability. Depending on the RBP, the stability can be increased or decreased significantly; however, miRNAs always decrease stability and promote decay.
9.16 Eukaryotic Translational and Post-translational Gene Regulation
Changing the status of the RNA or the protein itself can affect the amount of protein, the function of the protein, or how long it is found in the cell. To translate the protein, a protein initiator complex must assemble on the RNA. Modifications (such as phosphorylation) of proteins in this complex can prevent proper translation from occurring. Once a protein has been synthesized, it can be modified (phosphorylated, acetylated, methylated, or ubiquitinated). These post-translational modifications can greatly impact the stability, degradation, or function of the protein.
9.17 Cancer and Gene Regulation
Cancer can be described as a disease of altered gene expression. Changes at every level of eukaryotic gene expression can be detected in some form of cancer at some point in time. In order to understand how changes to gene expression can cause cancer, it is critical to understand how each stage of gene regulation works in normal cells. By understanding the mechanisms of control in normal, non-diseased cells, it will be easier for scientists to understand what goes wrong in disease states including complex ones like cancer.
9.18 Biotechnology
Nucleic acids can be isolated from cells for the purposes of further analysis by breaking open the cells and enzymatically destroying all other major macromolecules. Fragmented or whole chromosomes can be separated on the basis of size by gel electrophoresis. Short stretches of DNA or RNA can be amplified by PCR. Southern and northern blotting can be used to detect the presence of specific short sequences in a DNA or RNA sample. The term “cloning” may refer to cloning small DNA fragments (molecular cloning), cloning cell populations (cellular cloning), or cloning entire organisms
(reproductive cloning). Genetic testing is performed to identify disease-causing genes, and gene therapy is used to cure an inheritable disease.
Transgenic organisms possess DNA from a different species, usually generated by molecular cloning techniques. Vaccines, antibiotics, and hormones are examples of products obtained by recombinant DNA technology. Transgenic plants are usually created to improve characteristics of crop plants.
9.19 Mapping Genomes
Genome mapping is similar to solving a big, complicated puzzle with pieces of information coming from laboratories all over the world. Genetic maps provide an outline for the location of genes within a genome, and they estimate the distance between genes and genetic markers on the basis of recombination frequencies during meiosis. Physical maps provide detailed information about the physical distance between the genes. The most detailed information is available through sequence mapping. Information from all mapping and sequencing sources is combined to study an entire genome.
9.20 Whole-Genome Sequencing
Whole-genome sequencing is the latest available resource to treat genetic diseases. Some doctors are using whole-genome sequencing to save lives. Genomics has many industrial applications including biofuel development, agriculture, pharmaceuticals, and pollution control. The basic principle of all modern-day sequencing strategies involves the chain termination method of sequencing.
Although the human genome sequences provide key insights to medical professionals, researchers use whole-genome sequences of model organisms to better understand the genome of the species. Automation and the decreased cost of whole-genome sequencing may lead to personalized medicine in the future.
9.21 Applying Genomics
Imagination is the only barrier to the applicability of genomics. Genomics is being applied to most fields of biology; it is being used for personalized medicine, prediction of disease risks at an individual level, the study of drug interactions before the conduct of clinical trials, and the study of microorganisms in the environment as opposed to the laboratory. It is also being applied to developments such as the generation of new biofuels, genealogical assessment using mitochondria, advances in forensic science, and improvements in agriculture.
9.22 Genomics and Proteomics
Proteomics is the study of the entire set of proteins expressed by a given type of cell under certain environmental conditions. In a multicellular organism, different cell types will have different proteomes, and these will vary with changes in the environment. Unlike a genome, a proteome is dynamic and in constant flux, which makes it both more complicated and more useful than the knowledge of genomes alone.
Proteomics approaches rely on protein analysis; these techniques are constantly being upgraded. Proteomics has been used to study different types of cancer. Different biomarkers and protein signatures are being used to analyze each type of cancer. The future goal is to have a personalized treatment plan for each individual.
Adapted from:
OpenStax, Biology. OpenStax. May 20, 2013. <http://cnx.org/content/col11448/latest/>
“Download for free at http://cnx.org/content/col11448/latest/.”
